MEGA软件——系统发育树构建方法
- 格式:doc
- 大小:1.49 MB
- 文档页数:15
MEGA软件——系统发育树构建方法(图文讲解)
2012年12月02日⁄Evolution⁄字号小中大⁄评论 3 条⁄阅读 3,872 次[点击加入在线收藏夹]
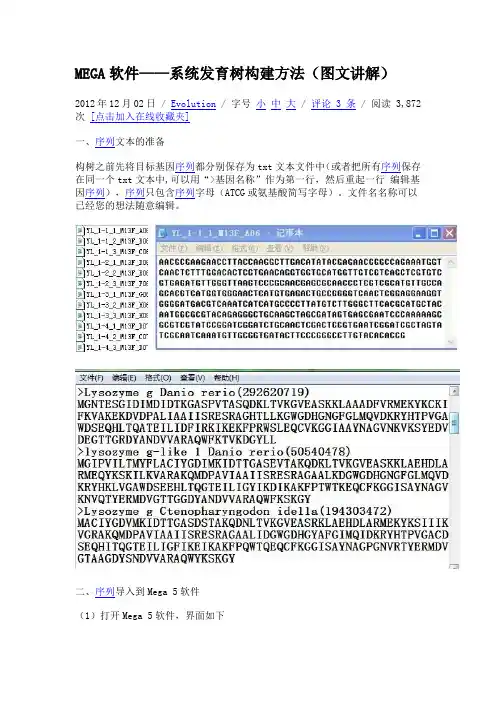
一、序列文本的准备
构树之前先将目标基因序列都分别保存为txt文本文件中(或者把所有序列保存在同一个txt文本中,可以用“>基因名称”作为第一行,然后重起一行编辑基因序列),序列只包含序列字母(ATCG或氨基酸简写字母)。
文件名名称可以已经您的想法随意编辑。
二、序列导入到Mega 5软件
(1)打开Mega 5软件,界面如下
(2)导入需要构建系统发育树的目的序列
OK
选择分析序列类型(如果是DNA序列,点击DNA,如果是蛋白序列,点击Protein)
出现新的对话框,创建新的数据文件
选择序列类型
导入序列
导入序列成功。
(3)序列比对分析
点击工具栏中“W”工具,进行比对分析,比对结束后删除两端不能够完全对齐碱基
(4)系统发育分析
关闭窗口,选择保存文件路径,自定义文件名称
三、系统发育树构建
根据不同分析目的,选择相应的分析算法,本例子以N—J算法为例
Bootstrap 选择1000,点击Compute,开始计算
计算完毕后,生成系统发育树。
根据不同目的,导出分析结果,进行简单的修饰,保存
本方法来自网络,经小编microibs编辑,修改补充,如果转载请注明PLoB出处。


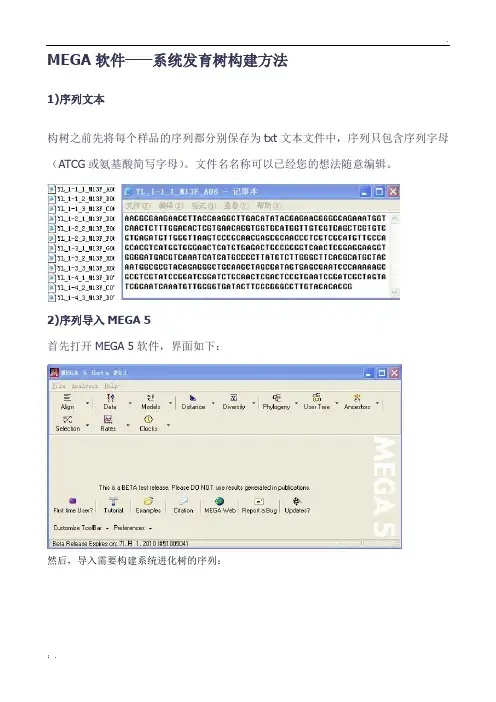
MEGA软件——系统发育树构建方法1)序列文本构树之前先将每个样品的序列都分别保存为txt文本文件中,序列只包含序列字母(ATCG或氨基酸简写字母)。
文件名名称可以已经您的想法随意编辑。
2)序列导入MEGA 5首先打开MEGA 5软件,界面如下:然后,导入需要构建系统进化树的序列:点击OK出现新的对话框,创建新的数据文件导入成功3)序列比对分析点击W,开始比对。
比对完成后删除序列两端不能完全对其的碱基。
系统分析然后,关闭该窗口,在弹出的对话框中选择保存文件,文件名随便去,比如保存为1。
4)系统发育树构建以NJ为例Bootstrap选择1000,点Computer,开始计算计算完毕后,生成系统发育树。
以下“系统发育树树的修饰”方法沿用斑竹brightfuture01的方法5)树的修饰建好树之后,往往需要对树做一些美化。
这个工作完全可以在word中完成,达到发表文章的要求。
点击image,copy to clipboard。
新建一个word文档,选择粘贴。
见下图:在图上点击右键-编辑图片,就可以对文字的字体大小,倾斜等做出修饰。
见下图:这个时候可以通过Adobe professional 对其进行图像导出:先将此word文档打印成PDF,见下图:将打印出来的PDF保存在桌面上,打开,如下图:此时,点击工具,高级编辑工具,裁剪工具,如下图所示:选择需要的区域以删除周围的空白区,双击发育树,会出现下图:点击确定,出现下图(把空边切掉了):点击文件,另存为,在保存类型一栏中选择TIFF格式,点击确定后会生成下面这个图片,所生成图片绝对可以满足文章的发表:OK,结束了,自己玩一把吧。
系统发育树
1.软件准备
DNAman、MEGA
2.序列文件转换格式
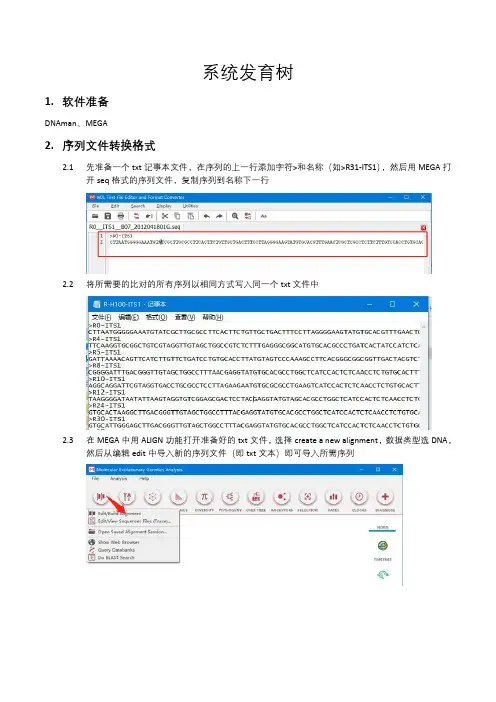
2.1先准备一个txt记事本文件,在序列的上一行添加字符>和名称(如>R31-ITS1),然后用MEGA打
开seq格式的序列文件,复制序列到名称下一行
2.2将所需要的比对的所有序列以相同方式写入同一个txt文件中
2.3在MEGA中用ALIGN功能打开准备好的txt文件,选择create a new alignment,数据类型选DNA,
然后从编辑edit中导入新的序列文件(即txt文本)即可导入所需序列
2.4删除无关序列后,先对序列进行分析然后再把序列对齐,类型选DNA,参数默认
颜色一致即为对齐,不一致的就是突变的位点。
然后通常需要把首尾两端没有对齐的序列删掉(只处理首尾两端未对其的序列)
对齐部分
未对齐的删掉
2.5处理完后保存文件并关闭当前窗口,如果不是连续使用的话,切换不同功能时一般点close date
关闭之前的数据
3.构建系统发育树
3.1邻接法构建系统发育树。

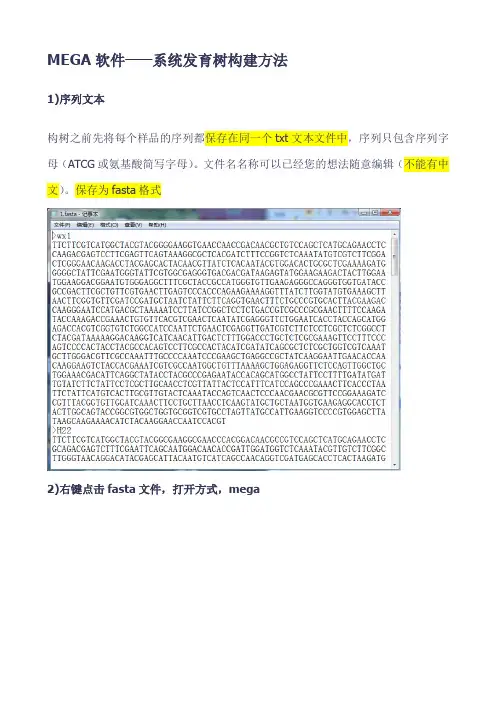
MEGA软件——系统发育树构建方法1)序列文本构树之前先将每个样品的序列都保存在同一个txt文本文件中,序列只包含序列字母(ATCG或氨基酸简写字母)。
文件名名称可以已经您的想法随意编辑(不能有中文)。
保存为fasta格式2)右键点击fasta文件,打开方式,mega3、全选,点击alignment,algin by culstx(按钮W),OK4、关闭此窗口,点击Yes保存5、再次点击Yes保存,6、点击cancel取消7、选择是否为编码蛋白质的核酸序列8、选择是否用mega打开文件9、点击YES,激活mega,此时mega的菜单栏与刚开始打开的菜单栏有区别。
10、系统发育树构建原理不讲了,此处以构建NJ树为例。
点击工具栏上的phylogeny,construct phylogeny,neighbor joining (NJ).出现如下界面(注意几个绿颜色的小方块):点击第一个小绿方块,选择,小绿方块会变成四个点的省略号,再点击出现如下页面:选择Bootstrap,后面的replication改为1000,点击对勾。
然后点击第三个小绿方块,这个时候对于蛋白质序列以及DNA序列,两者模型的选择是不同的。
对于蛋白质的序列,多选择Poisson Correction (泊松修正)这一模型。
而对于核酸序列,多选择Kimura 2-parameter (Kimura-2参数) 模型。
所有设置完毕之后,点击compute,雏形的树就出来了:可以对此树做出一些修改,比如线条粗细,树的形状等等,此处自己多试试。
6)树的修饰建好树之后,往往需要对树做一些美化。
这个工作完全可以在word中完成,达到发表文章的要求。
点击image,copy to clipboard。
新建一个word文档,选择粘贴。
见下图:在图上点击右键,就可以对文字的字体大小,倾斜等做出修饰。
见下图:PDF,见下图:将打印出来的PDF保存在桌面上,打开,如下图:此时,点击工具,高级编辑工具,裁剪工具,如下图所示:选择需要的区域以删除周围的空白区,双击发育树,会出现下图:点击确定,出现下图(把空边切掉了):点击文件,另存为,在保存类型一栏中选择TIFF格式,点击确定后会生成下面这个图片,所生成图片绝对可以满足文章的发表:OK,结束了,自己玩一把吧。

MEGA构建系统进化树的步骤(以MEGA7为例)本文是看中国慕课山东大学生物信息学课程总结出来的分子进化的研究对象是核酸和蛋白质序列。
研究某个基因的进化,是用它的DNA序列,还是翻译后的蛋白质序列呢?序列的选取要遵循以下原则:1)如果DNA序列的两两间的一致度≥70%,选用DNA 序列。
因为,如果DNA序列都如此相似,它的蛋白质会相似到看不出区别,这对构建系统发生树是不利的。
所以这种情况下应该选用DNA序列,而不选蛋白质序列。
2)如果DNA序列的两两间的一致度≤70%,DNA序列和蛋白质序列都可以选用。
1. 将要用于构建系统进化树的所有序列合并到同一个fasta格式文件,注意:所有序列的方向都要保持一致( 5’-3’)。
想要做系统发生树先要做多序列比对,然后把多序列比对的结果提交给建树软件进行建树,所以在用MEGA建树时可以输入一个已经比对好的多序列比对,也可以输入一条原始序列,让MEGA先来做多序列比对,再建树(一般我们都是原始序列)。
所以我们以后者为例。
2.打开MEGA软件,选择主窗口的”File”→“Open A File”→找到并打开fasta文件,这时会询问以何种方式打开,我们是原始序列,需要先进行多序列比对,所以选择“Align”。
如果是比对好的多序列比对可以直接选择“Analyze”。
3.在打开的Alignment Explorer窗口中选择”Alignment”-“Align by ClustalW”进行多序列比对(MEGA提供了ClustalW和Muscle两种多序列比对方法,这里选择熟悉的ClustalW),弹出窗口询问“Nothing selected for alignment,Select all?”选择“OK”。
4. 之后,弹出多序列比对参数设置窗口。
这个窗口和EMBL在线多序列比对一样,可以设置替换记分矩阵、不同的空位罚分(罚分填写的是正数,计算时按负数计算)等参数。

MEGA构建系统进化树的步骤(以MEGA7为例)本文是看中国慕课山东大学生物信息学课程总结出来的分子进化的研究对象是核酸和蛋白质序列。
研究某个基因的进化,是用它的DNA序列,还是翻译后的蛋白质序列呢?序列的选取要遵循以下原则:1)如果DNA序列的两两间的一致度≥70%,选用DNA 序列。
因为,如果DNA序列都如此相似,它的蛋白质会相似到看不出区别,这对构建系统发生树是不利的。
所以这种情况下应该选用DNA序列,而不选蛋白质序列。
2)如果DNA序列的两两间的一致度≤70%,DNA序列和蛋白质序列都可以选用。
1. 将要用于构建系统进化树的所有序列合并到同一个fasta格式文件,注意:所有序列的方向都要保持一致( 5’-3’)。
想要做系统发生树先要做多序列比对,然后把多序列比对的结果提交给建树软件进行建树,所以在用MEGA建树时可以输入一个已经比对好的多序列比对,也可以输入一条原始序列,让MEGA先来做多序列比对,再建树(一般我们都是原始序列)。
所以我们以后者为例。
2.打开MEGA软件,选择主窗口的”File”→“Open A File”→找到并打开fasta文件,这时会询问以何种方式打开,我们是原始序列,需要先进行多序列比对,所以选择“Align”。
如果是比对好的多序列比对可以直接选择“Analyze”。
3.在打开的Alignment Explorer窗口中选择”Alignment”-“Align by ClustalW”进行多序列比对(MEGA提供了ClustalW和Muscle两种多序列比对方法,这里选择熟悉的ClustalW),弹出窗口询问“Nothing selected for alignment,Select all?”选择“OK”。
4. 之后,弹出多序列比对参数设置窗口。
这个窗口和EMBL在线多序列比对一样,可以设置替换记分矩阵、不同的空位罚分(罚分填写的是正数,计算时按负数计算)等参数。

利⽤MEGA-X选择模型及构建美化进化树今天主要介绍的是在MEGA-X图形界⾯下构建系统发育树并且对发育树进⾏美化。
下载安装好MEGA-X后,⾸先打开软件。
此处我们以⼀株细菌的16S rRNA序列为⽬标序列,⾸先在NCBI中进⾏Blast⽐对,下载将要⼀起⽐对建树的菌株序列。
在NCBI中输⼊序列或者上传⽂件,选择数据库时可以选择「Nucleotide collection(nr/nt)」或者「16S ribosomal RNA sequences」数据库,⼀般来说nr/nt库信息⽐较全⾯。
我们选择了10个不同种的16S rRNA序列进⾏下载。
另外,此处还可以⽐对下载2-3条⼤肠杆菌(Escherichia coli)和沙门⽒杆菌(Salmonella)的16S rRNA序列作为外类群(在Organism选项中进⾏物种限定),后⾯推断进化时间的时候可以⽤到。
将所有下载的序列整理在⼀个⽂件中,为了⽅便后⾯的建树可以将菌株名称后⾯多余的信息在这⾥替换删除掉(只是名称上的信息,不要改动碱基序列),然后将⽂件的扩展名改为.fasta。
在MEGA-X⾸页选择DATA,点击Open a File/Session,选择刚才的⽂件。
打开⽂件时询问「Analyze or Align File?」,此处点击Align。
序列中可能会出现混合碱基符号,混合碱基符号指两种或多种碱基(核苷)混合物的表⽰符号,或未完全确定可能属于某两种或多种碱基(核苷)的符号:R表⽰A+G;Y表⽰C+T;M表⽰A+C;K表⽰G+T;S表⽰C+G;W 表⽰A+T;H表⽰A+C+T;B表⽰C+G+T;V表⽰A+C+G;D表⽰A+G+T;N表⽰A+C+G+T。
接下来选择序列⽐对的⽅法:Muscle或者ClustalW。
ClustalW的基本原理是⾸先做序列的两两⽐对,根据该两两⽐对计算两两距离矩阵,是⼀种经典的⽐对⽅法,使⽤范围也⽐较⼴泛。
Muscle的功能仅限于多序列⽐对,它的最⼤优势是速度,⽐ClustalW的速度快⼏个数量级,⽽且序列数越多速度的差别越⼤。
使用MEGA6.0软件构建系统发育树所有序列可以在Bioedit里整理成为一个FASTA文件将FASTA格式的文件在MEGA软件里打开123点击Open A File/Session,打开FASTA格式的文件4点击Align56点击Edit,Select All7点击W图标,或者8 点击OK(默认参数)910 删除头尾有空缺的地方11 点击Data,选择phylogenetic Analysis,将这个窗口最小化12 MEGA软件窗口增加了两个方框13 点击Phylogeny,选择需要建树的类型,以NJ树为例14 询问是否使用当前数据继续建树,点击Yes15 数值设置好后,点击Compute16 表示程序正在运行17 原始结果显示如下,可以根据自己的需求进行调整18 View里可以调整数值宽度等1920 图片导出,点击Image,选择Copy to Clipboard,粘贴到word文档2121 选中图片,点击编辑图片,对字体大小及内容能够进行调整与修改AAG51164.1Arabidopsis thalianaAAL35328.1 Oryza sativ aCAA43142.1 Malus domesticaNP 001281081.1 Zea maysGAQ91141.1Klebsormidium flaccidumAAA34144.1 Solanum lycopersicumADD85140.1 Triticum aestiv umNP 031615.1 Mus musculusNP 001286337.1Drosophila melanogaster AES82664.1 Medicago truncatulaCAA55612.1 Saccharomyces cerev isiae。
系统发育分析-MEGA实 验 目 的1. 学会使用 MEGA 构建进化树,熟悉建树相关参数;2. 会分析建树结果,体会不同方法的差异。
实 验 内 容 实 验 流 程一、 准备工作首先现在MEGA 的官网上下载MEGA X :正式下载前还需要输入一些信息:在宿舍用Wi-Fi 下载也是极慢(1M/min ),用VPN1分钟就直接下好了,安装:由于我们之前的同源序列中只有直系同源序列,因此我们需要再在序列库中寻找MTPAP的并系同源序列。
首先在NCBI的HomoloGene库中搜索MTPAP,得到以下结果:点击Orthologs,可以发现许多的直系同源基因,由于之前选择的5条序列相似度过高,因此我们重新下载10条直系同源核酸与蛋白序列:发现了一个从未听说过,但功能却极为强大的网站——GeneCard:信息:所有序列整理好后如下图所示:在此之前,先将物种名单信息上传至NCBI的Taxonomy - Common Tree中,找到要与建树结果比对的标准树:使用TreeViewX打开下载得到的phy文件:可以看到,重新下载的物种数据分布比较宽泛,避免因序列信息过于相似而使建树结果出现分歧。
二、直系同源序列的比对现在正式开始使用MEGA X进行序列分析。
先利用MEGA的多序列比对功能得到meg文件。
导入序列:使用ClustalW进行比对:比对结果:保存比对结果(可以选择fasta格式或是meg格式)。
三、对五种建树方法的探索我们分别尝试五种建树方法,并于标准树做对比:1. ML法建树参数设置:Original ML Tree:100次Bootstrap后得到的一致树:可以看到,Bootstrap前后,直系同源序列的关系都与是否与已知物种分类关系相同,只是拓扑结构略有区别。
2. NJ法建树参数设置:Original NJ Tree:100次Bootstrap后得到的一致树:相对于ML法,NJ法建树速度极快,但是建树结果就是在是差强人意了。
图文详解MEGA 5构建系统发育树(2013-10-11 20:52:49)如遇不妥,请指正。
软件下载:MEGA 5 ; DNAMAN 71 •准备序列文件准备fasta格式序列文件(fasta格式:大于号> 后紧跟序列名,换行后是序列。
举例如下)。
每条序列可以单独为一个文件,也可以把所有序列放在同一文件内。
核酸序列:>sequence1_nameCCTGGCTCAGGATGAACGCT氨基酸序列:>sequence2_nameMQSPINSFKKALAEGRTQIGF2 .多序列比对打开MEGA 5,点击Align,选择Edit/Build Alignment ,选择Create a new alignment ,点击OK。
MEGA 5,05File Analysis HelpOpen Saved Alignment Session,..玄Show Web Browser& Query DstabankEDo BLAST SearchSelect an Option -■ * Create a new dig nment( Open a saved alignment sessiDn「Retrieve sequencer from a file4 OK X Parcel这时需要选择序列类型,核酸(DNA )或氨基酸(Protein )选择之后,在弹出的窗口中直接 Ctrl + V 粘贴序列(如果所有序列在同一 个文件中,即可全选序列,复制)。
也可以:点击 Edit ,选择Insert Seque nee From File ,选择序列文件(可多选)。
TA■I 鼻■ •DataModefcAl 卯Edrt/View £equ>ercer Ries (Tracc)^Edrt/Build AlignmentDistancM5: Aligrrnnent Editor© S3(为选中状态)。
系统进化树制作步骤MEGA5.1
先要把格式弄成该软件识别的meg格式,fasta格式也行,只要能够导入
1导入,点击左上角Align,选择创建新的比对Alignment,点击OK,
2提示创建DNA分析文件,进入如下右边界面
3打开fasta格式的序列文件,如下
4选择Alignment中的比对选项,使用Clastaw比对,会有比对参数,默认即可,点OK,会自行完成比对。
5比对好的文件须保存,选Data中的save session,提示保存命名
即导入数据如右。
7选中带有TA的方框,如上图,点击主界面上的进化树选项,如下图中间,可选择不同的
进化树类型,一般用NJ树。
8出现提示参数,默认即可,点compute执行。
会完成进化树
9做好的进化树可以以图片方式保存。
MEGA软件——系统发育树构建方法
1)序列文本
构树之前先将每个样品的序列都分别保存为txt文本文件中,序列只包含序列字母(ATCG或氨基酸简写字母)。
文件名名称可以已经您的想法随意编辑。
2)序列导入MEGA 5
首先打开MEGA 5软件,界面如下:
然后,导入需要构建系统进化树的序列:
点击OK
出现新的对话框,创建新的数据文件
导入成功
3)序列比对分析
点击W,开始比对。
比对完成后删除序列两端不能完全对其的碱基。
系统分析然后,关闭该窗口,在弹出的对话框中选择保存文件,文件名随便去,比如保存为1。
4)系统发育树构建
以NJ为例
Bootstrap选择1000,点Computer,开始计算
计算完毕后,生成系统发育树。
以下“系统发育树树的修饰”方法沿用斑竹brightfuture01的方法
5)树的修饰
建好树之后,往往需要对树做一些美化。
这个工作完全可以在word中完成,达到发表文章的要求。
点击image,copy to clipboard。
新建一个word文档,选择粘贴。
见下图:
在图上点击右键-编辑图片,就可以对文字的字体大小,倾斜等做出修饰。
见下图:
这个时候可以通过Adobe professional 对其进行图像导出:先将此word文档打印成PDF,见下图:
将打印出来的PDF保存在桌面上,打开,如下图:
此时,点击工具,高级编辑工具,裁剪工具,如下图所示:
选择需要的区域以删除周围的空白区,双击发育树,会出现下图:
点击确定,出现下图 (把空边切掉了):
点击文件,另存为,在保存类型一栏中选择 TIFF格式,点击确定后会生成下面这个图片,所生成图片绝对可以满足文章的发表:
OK,结束了,自己玩一把吧。
如有侵权请联系告知删除,感谢你们的配合!。