群体重测序与大山雀的群体进化
- 格式:pdf
- 大小:14.49 MB
- 文档页数:2

不同种类生物的分类与进化生物的分类和进化是生物学中非常重要的一部分。
生物在进化过程中形成了各种形态、习性和适应性,这些都是因为它们所处的环境不断地变化。
而在分类学中,生物们被分为不同的类别,使得大家能够更好地理解它们之间的关系。
在本文中,我们将会深入探讨生物的分类和进化这个主题。
分类学是一个将生物进行分类的科学。
这个学科在生物发展史上非常重要,因为它使得人类能够更好地理解和研究生物。
所有的生物都按照它们的共同特征和遗传关系来进行分类。
按照这个规则,生物可以被分为不同的层级,包括:物种、属、目、纲、门和界。
这些层级之间形成了一种分类法,被称为生物分类学。
当生物被归类时,物种是分类法的基石。
生物学家们采用典型的形态、习性和分子遗传学等多种方法来鉴定物种。
例如,同一物种的所有个体都具有类似的形态、结构和组成,而不同物种的个体则会有某些显著区别。
物种的鉴定使得生物学家们可以确定生物的数量和范围。
在分类学中,生物可以按照它们的亲缘关系被分成不同的类别。
这些类别都有着不同的名称和特征。
比如,一组共享同一特征的物种就被划分成一个属。
然后,各个属可以再被划分到同一个目中,同一个目中的各个属再被划分到同一个纲中。
如此重复下去,直至形成了完整的生物分类学。
然而,分类学并不是一成不变的,生物的分类也是在不断发生变化的。
由于新技术和新证据的出现,生物学家们不断地对生物进行重新分类。
例如,基因测序可以探究生物的分子结构,这使得分类学家们可以通过分析基因序列的差异来鉴定物种之间的联系。
同样的,生物学家们也会重新审视已有物种的分类,也会发现新的物种以及它们之间的关系。
这些变化不仅会让生物分类学更加准确,同时也会让我们对生物进化的理解更加深入。
分类学与生物进化息息相关。
进化是指生物在遗传和遗传环境中逐步变化,让它们能够更好地适应环境。
在进化过程中,生物会相互竞争,也会相互依存。
通过进化,生物得以适应其所居住的环境,并且能够在不断变化的生态系统中生存下来。



生物大数据技术中的群体遗传结构分析方法介绍随着生物信息学和基因测序技术的快速发展,我们现在可以收集到大量的生物数据。
这些大数据有助于我们理解物种内部的遗传变异和群体遗传结构。
群体遗传结构分析是研究同一物种群体内不同个体之间的遗传联系与差异的一种重要方法。
在这篇文章中,我们将介绍几种常用的群体遗传结构分析方法。
1. 群体结构分析(Population Structure Analysis)群体结构分析是通过分析群体内不同个体间的遗传差异,将它们分成几个亚群体或种群。
这种方法可以用来研究物种内部的亚种分化或者种群间的迁移情况。
其中最常用的方法是主成分分析(Principal Component Analysis, PCA)。
主成分分析可以将多个遗传变异指标进行降维处理,帮助我们发现潜在的群体结构。
2. 迁移率和交配模式推断(Migration and Mating Pattern Inference)迁移率和交配模式推断是研究群体遗传结构演化过程中迁移率和交配模式的一种方法。
在某些物种中,不同种群之间的迁移率对于维持物种的遗传多样性和适应性具有重要意义。
而交配模式则可以告诉我们遗传信息是如何在不同个体之间进行交换的,从而揭示了物种内部的基因流动情况。
常用的分析方法包括STRUCTURE和TASSEL等软件。
3. 基因流动分析(Gene Flow Analysis)基因流动分析是研究不同地理或种群间的基因交流情况的一种方法。
基因流动是指不同群体或种群之间的基因交换。
通过分析个体间的遗传联系,我们可以推断基因流动的程度和方向。
这对于研究物种的分布、扩散和适应性非常重要。
常用的方法包括DAPC和STRUCTURE等。
4. 遗传分化度量(Genetic Differentiation Measure)遗传分化度量是用来衡量不同种群间遗传差异的一种方法。
通过计算不同种群间的遗传距离、遗传分化系数或遗传差异指数,我们可以了解不同种群之间的遗传关系。

重测序bsa技术原理重测序BSA技术原理是现代遗传学中最重要的技术之一。
它是一种通过环状PCR扩增技术和测序高通量技术检测基因变异的方法。
本文将从技术原理、方法流程、优缺点等方面详细阐述。
首先,BSA技术原理基于遗传位点在杂交种群中的分离,通过PCR扩增、库建立、测序等一系列操作,筛选出突变位点。
具体步骤如下:第一步,制备DNA样品。
需要从不同菌株或植株样本中提取DNA 样品,纯化后合并成同等质量的混合DNA样品。
第二步,BSA过滤。
将混合DNA样品进行PCR扩增,得到一条长约300-500bp的PCR产物。
然后进行BSA过滤,筛选出对应位点的PCR 条带,进行回收。
第三步,建立DNA文库。
回收的PCR产物首先要进行文库建立。
文库的建立方法有很多种,最常用的是同时使用T4 DNA聚合酶和多个ATP酰化酶,将PCR产物扩增并文库化。
建立好的文库可以用于后续测序分析。
第四步,高通量测序。
测序可以使用目前流行的Illumina测序平台。
测序结果会生成一系列序列片段,我们可以利用这些片段进行序列比对和SNP鉴定等分析。
第五步,SNP鉴定。
利用比对软件将测序片段与原来基因组数据进行比对,鉴定出SNP突变位点,进一步进行验证和鉴定。
通过以上一系列操作,我们可以在遗传群体内筛选出突变位点,并判断是否遗传贡献存在,从而进一步研究与分析这种遗传变异的功能和鉴定路径等问题。
BSA技术相对于其他遗传分析方法有着明显的优点。
首先,它可以针对各种生物材料,不分样品来源与类型;其次,它具有很高的分辨率。
在物种较少的情况下,BSA技术可以直接对突变位点进行鉴定并验证,从而保证了分析的准确性;同时,BSA技术的重点在于检测遗传双一体性,比起散点分析等其他分析手段更加容易分析遗传变异。
但是,BSA方法也有一些缺点。
首先,BSA技术在筛选突变位点方面存在一定的局限性。
如果突变在杂交种群中的频率过低、过高或存在不同的等位基因则会干扰筛选的准确性。

基因组重测序
基因组重测序(Genome Resequencing)是一种研究族群遗传学和物种进化过程的常用分析方法,它包括对个体或物种基因组的重新测序,以及对基因组的遗传变异的进一步探讨。
基因组重测序可以用来研究物种进化,筛选便利性基因以及鉴定和分析基因组变异。
一、优势
1、基因组重测序的比较优势:重测序比利用芯片进行平面分析方法更加灵活。
能够快速鉴定多种类型的遗传变异,包括插入、缺失、临时变异,以及双倍体变异等。
2、复杂性大:由于重测序可以精细分析基因组中的染色体,因此可以更好地捕捉基因组变异的复杂性。
3、高效性:仪器分析周期短,该技术可以高效地获得基因组芯片和组装基因组变异的信息。
二、应用
1、种群遗传研究:基因组重测序能够针对个体或物种基因组的群体变异和单倍型进行分析,以发现先前未被准确定位的遗传标记和位点,有助于预测物种进入新环境时适应性和抗病性方面的变异。
2、育种研究:基因组重测序可以鉴定出品质和适应性相关的基因和位点,有助于精准育种。
3、公共健康:基因组重测序可以确定某种疾病的发病形态,有助于进
一步深入认识疾病的发生机理以及发病的根源,从而促进公共健康的发展。
三、前景
在未来,基因组重测序技术将会被广泛应用于基因组学中,例如用于进化生物学和疾病基因组学研究,它也可用于转基因技术和育种。
同时也会继续发展新的基因组重测序技术,更新、完善重测序技术,为科学家和科技工作者提供更多先进的应用技术。

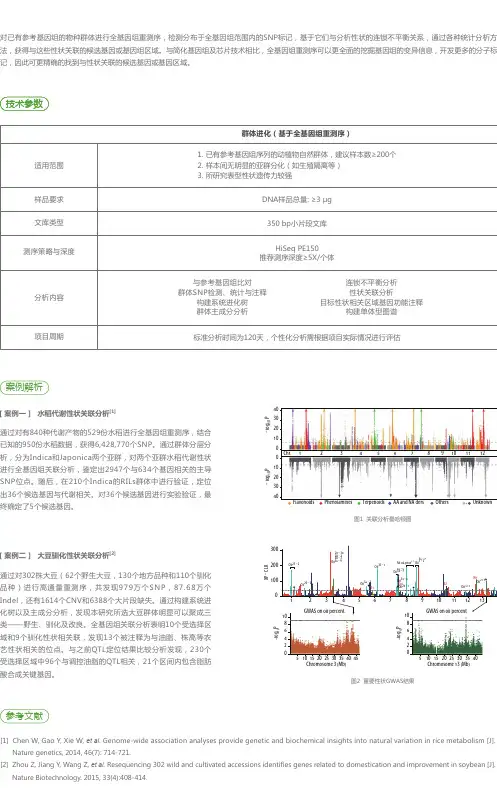
DNA样品总量: ≥3 μg 适用范围样品要求文库类型测序策略与深度分析内容项目周期 群体进化(基于全基因组重测序)标准分析时间为120天,个性化分析需根据项目实际情况进行评估HiSeq PE150推荐测序深度≥5X/个体350 bp小片段DNA文库1. 已有参考基因组序列的物种中不同亚群(自然群体)2. 各亚群间划分明显,同一亚群内的个体有一定代表性3. 每个亚群选取10个样本左右(推荐动物≥10个,植物≥15个)4. 总体不少于30个样本与参考基因组比对群体SNP检测、注释及统计系统进化树构建群体遗传结构分析群体主成分分析连锁不平衡分析选择消除分析候选基因GO和KEGG富集构建单体型图谱种群历史和有效群体大小技术参数针对已有参考基因组的物种,对其各亚种进行全基因组重测序获得基因组信息,通过与参考基因组比对,得到大量高准确性的SNP、InDel、SV等变异信息,讨论群体的遗传结构、遗传平衡和影响遗传平衡的因素,从而从分子层面揭示该物种的进化机制、环境适应性等系列问题。
该技术能精准地得到全基因组内所有遗传信息,最大程度地挖掘出群体内遗传变异。
诺禾具有丰富的群体遗传学项目经验,研究成果发表于Nature Genetics(Li, M, et al. 2013& Zhou, XM,et al. 2014)等。
参考文献[1] Li M, Tian S, Jin L, et al . Genomic analyses identify distinct patterns of selection in domesticated pigs and Tibetan wild boars [J]. Nature genetics, 2013, 45(12): 1431-1438.[2] Zhan S, Zhang W, Niitepo ~ld K, et al . The genetics of monarch butterfly migration and warning colouration [J]. Nature, 2014.案例解析[案例一] 家猪和藏猪的群体进化分析[1]2013年,诺禾致源科技服务团队与四川农业大学研究者合作发表该成果。

鸟类生态学的研究方法鸟类生态学是对鸟类及其生态环境进行科学研究的学科。
通过对鸟类的生境利用、种群动态、社会行为等方面进行观察和分析,揭示鸟类与环境的相互作用关系。
为进行鸟类生态学的研究,需要采用一系列科学的方法和技术工具。
本文将介绍鸟类生态学的研究方法,包括现场观察、标记重捕法、遥感技术等。
一、现场观察法现场观察法是鸟类生态学中最常用、最基础的研究方法之一。
研究人员在鸟类常出没的地区设立固定观测点,通过直接观察记录鸟类的种类、数量、行为等信息。
这项工作需要研究人员具备丰富的鸟类识别经验和观察技巧,同时需要耐心和细致,以保证数据的准确性和可靠性。
二、标记重捕法标记重捕法是一种常见的鸟类生态学研究方法,通过对部分鸟类个体进行标记并对其进行定期抓捕、重复观察,来研究种群的生长、死亡、迁移等参数。
常见的标记方法包括足环、颈环、羽毛剪裁等,研究人员通过观察和记录标记鸟个体的状况变化,结合数学模型进行数据分析和预测。
三、遥感技术遥感技术是近年来在鸟类生态学研究中得到广泛应用的一种方法。
通过利用航空或卫星遥感图像,研究人员可以获取广大范围内的环境数据,包括植被覆盖、地形变化等。
这些数据可以与鸟类的分布、迁徙等信息进行对比和分析,揭示鸟类与环境的相互关系。
遥感技术不仅可以提供大范围的信息,还可以反复观察以获取时间序列的数据,有利于长期趋势和变化的研究。
四、分子生态学方法分子生态学是鸟类生态学中兴起的一种新的研究方法。
通过分析鸟类DNA或RNA的序列数据,可以揭示种群的遗传结构、群体间的遗传流动、种群的进化历史等信息。
分子生态学方法可以辅助传统的观察和实验,提供更详细的鸟类生态学信息。
五、模型和统计分析在鸟类生态学的研究中,模型和统计分析是不可或缺的工具。
通过搜集和整理大量的观测数据,研究人员可以建立数学模型,并利用统计方法对数据进行分析和解释。
模型和统计分析可以帮助研究人员验证假设、预测趋势、发现规律,对鸟类生态学研究的科学性和可靠性起到重要的支撑作用。
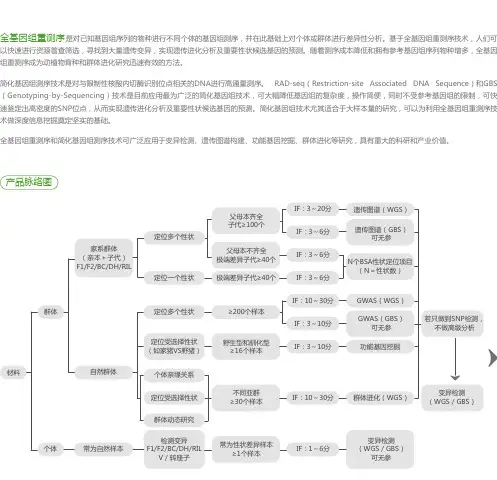
全基因组重测序是对已知基因组序列的物种进行不同个体的基因组测序,并在此基础上对个体或群体进行差异性分析。
基于全基因组重测序技术,人们可以快速进行资源普查筛选,寻找到大量遗传变异,实现遗传进化分析及重要性状候选基因的预测。
随着测序成本降低和拥有参考基因组序列物种增多,全基因组重测序成为动植物育种和群体进化研究迅速有效的方法。
简化基因组测序技术是对与限制性核酸内切酶识别位点相关的DNA进行高通量测序。
RAD-seq(Restriction-site Associated DNA Sequence)和GBS (Genotyping-by-Sequencing)技术是目前应用最为广泛的简化基因组技术,可大幅降低基因组的复杂度,操作简便,同时不受参考基因组的限制,可快速鉴定出高密度的SNP位点,从而实现遗传进化分析及重要性状候选基因的预测。
简化基因组技术尤其适合于大样本量的研究,可以为利用全基因组重测序技术做深度信息挖掘奠定坚实的基础。
全基因组重测序和简化基因组测序技术可广泛应用于变异检测、遗传图谱构建、功能基因挖掘、群体进化等研究,具有重大的科研和产业价值。
产品脉络图。

辽宁大学学报自然科学版第40卷第3期2013年J O U R N A L O F L I A oN I N G U NⅣE R s nYN at u r al Sci ences E出t i onV0l。
40N o.32013中国大陆杂色山雀遗传多样性万冬梅+,鞠静,蔡碉,张雷,李东来(辽宁大学生命科学院辽宁省动物资源与疫病防治重点实验室,辽宁沈阳110036)摘要:使用线粒体控制1爰(529bp)和核基因(9个微卫星位点)作为分子标记对杂色山雀(Parus var i us)在中国大陆8个分布地的70个体进行遗传多样性研究.在所有样本线粒体控制区序列中共识别出10个单倍型,整体核苷酸多样性(Pi)为0.00176;单倍型多样性(H a)为0.533;平均核苷酸差异数(k)为0.892.在线粒体水平上,杂色山雀中国大陆种群处于线粒体D N A多态性较低的范围内,遗传多样性较低.微卫星数据显示,70个样本在9个微卫星位点上的多态信息含量(P/C)为0.305~0.863,平均多态信息含量为0.755±0.1720;8个分布地样本各自平均等位基因个数(置)在1.4±O.53—9.4.4-2.46之间,平均期望杂合度(^k)在0.4444-0.5270~o.∞8±o'1929之间,平均多态信息含量在0.167±O.1976—0.738±0.1634之间.在核基因水平上,杂色山雀中国大陆种群遗传信息丰富,遗传多样性较高.基于线粒体控制区序列构建的系统进化树显示中国大陆地区杂色山雀指名亚种8个分布地样本混杂在一起;样本间的遗传分化指数Fst为0.0119,基因交流值为3.82.样本中的大部分变异是来自种群内且这种变异达到遗传分化的显著性水平,显示了中国大陆杂色山雀指名亚种8个分布地样本为一个地理种群.错配分布图以及中性检验结果显示中国大陆杂色山雀在过去的发展历程中发生过扩张事件.对于中国大陆地区杂色山雀的保护,应该将其作为一个进化显著单元进行保护.关键词:杂色山雀;线粒体D N A(D—l oop);微卫星标记;遗传多样性中圈分类号:Q958文献标志码:A文章编号:1000-5846(2013)03-0249—11G enet i c D i ver si t y of Par us var i us i n M ai nl and of C hi naW A N D ong—m e i’,J U J i ng,C A I Y ue,Z H A N G L ei,L I D ong-l a i (K ey Labor a t or y of A ni m al R esource and Epi dem i c D i s e as e P r e vent i on,D e par t m ent of Li f e Sc i enc es,L i a oni ng U ni ve r si t y,Shenyang110036,C hi na)A bs t r act:T he l evel of genet i c di ver s i t y of speci al spe ci es i s vi t al t o i t s evol ut i onar y pot ent i al and ada pt at i on t o envi r onm ent change.To quant i f y t he genet i c di ver s i t y of Par us va r i us,w e exam i ned var i at i ons i n m i t ochondr i a l D N A(m t D N A)cont r ol r egi on(529bp)i n70i ndi vi dual s f rom8r epr ese nt at i ve di s t ri but i ons t hr oughout t he spec i e s’r a nge i n C hi na.I n addi t i on,al l i ndi vi dual s w er e genot yped w i t h9m i c r os am l l i m l oci.T e n ha pl ot ype s w ere de f i ned by14var i abl e收稿日期:2013一04一17基金项目:国家自然科学基金项目(31071927,30670288)作者简介:冬梅(1969一),女。
2024北京高三二模生物汇编遗传与进化(非选择题)一、非选择题1.(2024北京东城高三二模)铝毒害会限制植物生长,对农业和生态安全造成威胁。
对植物如何感知铝进而启动抗铝响应开展研究。
(1)无机盐在细胞中大多数以形式存在,对细胞和生物体的生命活动有重要作用。
有些无机盐对细胞有毒害,具有抗性的植物有更多的机会产生后代,经过长期的,后代抗性不断增强。
(2)为研究ALR1与植物抗铝性的关系,研究者利用拟南芥进行实验,测量根长并计算相对值,根长相对值=有金属离子处理的根长/无金属离子处理的根长×100%,结果如图1、图2。
综合图1、图2结果,推测。
(3)植物根分泌的有机酸阴离子能结合并限制铝离子进入根,这是植物抗铝性的核心作用。
ALMT1和MATE为有机酸盐外排转运蛋白,在条件下分别检测野生型、ALR1缺失突变体和ALR1过表达突变体植株根部细胞中相应基因的表达情况,结果表明这两种蛋白参与ALR1介导的抗铝性。
(4)ALMT1和MATE的表达由转录调控因子STOP1控制。
检测各组植株中STOP1的mRNA和蛋白含量,由此推测ALR1仅通过抑制STOP1蛋白水解调控植物的抗铝性。
请在图3中画出有铝条件下各组的实验结果。
(5)最终确定ALR1是铝离子受体。
除本题中提到的ALR1调控植物抗铝性的信号通路外,还需证明ALR1能,才能得出此结论。
2.(2024北京昌平高三二模)褐飞虱和白背飞虱是两种为害水稻的稻飞虱,研究者对二者的种间关系进行了一系列的研究。
(1)褐飞虱和白背飞虱都是通过吸食水稻汁液和在稻株上产卵等方式为害水稻,二者之间存在关系。
(2)水稻自身可释放挥发物,会影响褐飞虱和白背飞虱的行为。
研究者使用下图所示的“H”型嗅觉仪进行实验。
注:“H”型透明封闭装置中,A区放置稻飞虱,B和C区放置不同的水稻苗或土壤①实验中水稻挥发物作为物质发挥作用。
①在“H”型装置的B和C区放置不同的水稻苗或土壤,A区放置稻飞虱,一段时间后统计两侧的稻飞虱数量,为避免稻飞虱迁飞的影响,可通过法进行计数。
二代测序在遗传突变中的应用遗传突变是指在基因或染色体结构中发生的变异,它是生物进化和种群遗传多样性的重要驱动力,也是导致许多遗传疾病和肿瘤发生发展的根本原因。
随着二代测序技术(也称为高通量测序技术)的迅速发展,我们能够对遗传突变进行更为全面和精确的研究,以探索其在个体和种群水平上的重要性。
本文将介绍二代测序在遗传突变检测、疾病表型关联、肿瘤进化和个体遗传变异研究中的应用,并探讨其未来的发展方向。
一、遗传突变检测二代测序技术已经成为检测遗传突变最为常用的方法之一。
通过对个体基因组的全序列测定或目标区域的深度覆盖测序,我们可以发现一些单碱基变异、插入缺失、复杂重排等突变。
这些突变可能是导致遗传疾病发生的原因,也可能是个体遗传差异的来源。
二代测序技术的高通量和高灵敏性使得我们能够更全面地了解个体的遗传异质性,从而实现个性化医学的目标。
此外,二代测序还可以检测外源性核酸(如病毒、细菌等)的感染,并跟踪其在个体中的传播和演化。
二、疾病表型关联遗传突变与疾病表型之间存在着密切的关联。
通过对大样本群体的基因组测序和表型数据的整合分析,我们可以鉴定出与疾病发生发展有关的遗传变异。
这些变异可能是单基因遗传病的致病突变、复杂疾病的易感变异、基因表达调控元件的变异等。
二代测序技术的高通量能力使得我们能够在大样本群体中快速发现和验证这些与疾病相关的突变,为疾病的早期预测和个体化治疗提供了重要的依据。
三、肿瘤进化研究肿瘤是由遗传突变引起的一类疾病,肿瘤细胞在不断的演化过程中会积累大量的突变。
二代测序技术的出现为肿瘤进化和肿瘤异质性研究提供了无与伦比的机会。
通过对肿瘤样本和正常样本的基因组测序和比较分析,我们可以追踪肿瘤的起源、进化和扩散过程,揭示肿瘤的发展规律和临床表现的多样性。
此外,通过定向测序和组学数据的整合分析,我们也可以发现对特定药物敏感或抵抗的突变,并为肿瘤个体化治疗提供理论依据。
四、个体遗传变异研究每个个体都存在着独特的遗传变异,这些变异会影响个体的生理特性、药物反应以及疾病易感性等。
全基因组重测序项目简介全基因组重测序是对已有参考序列(Reference Sequence)的物种的不同个体进行基因组测序,并以此为基础进行个体或群体水平的差异性分析。
通过这种方法,可以寻找出大量的单核苷酸多态性位点(SNP),插入缺失位点(InDel,Insertion Deletion),结构变异位点(SV,Structure Variation),拷贝数变异(Copy Number Variation,CNV)等变异信息,从而获得生物群体的遗传特征。
这对在群体水平上研究物种的进化历史、环境适应性、自然选择等方面具有重大意义。
利用全基因组重测序有助于快速发现与动植物重要性状相关的遗传变异,缩短分子育种的实验周期;有助于发现人类疾病相关的重要变异基因,加快生物医药研发的速度等,这对人类疾病及动植物育种研究等方面具有重大的指导意义。
技术流程提取基因组DNA后,采用物理方法随机打断,选择性回收所需长度的DNA片段(0.2~5Kb),并在两端连接接头以构建测序文库,进行桥式PCR(Bridge Amplification)制备Cluster,最后利用Paired-End的方法对插入片段进行重测序。
生物信息分析1.数据量产出总碱基数量、Totally mapped reads、Uniquely mapped reads统计,测序深度分析。
2.一致性序列组装与参考基因组序列(Reference genome sequence)的比对分析,利用贝叶斯统计模型检测出每个碱基位点的最大可能性基因型,并组装出该个体基因组的一致序列。
3.SNP检测及在基因组中的分布提取全基因组中所有多态性位点,结合质量值、测序深度、重复性等因素作进一步的过滤筛选,最终得到可信度高的SNP数据集。
并根据参考基因组序列对检测到的变异进行注释。
4.InDel检测及在基因组的分布在进行mapping的过程中,进行容Gap的比对并检测可信的Short InDel。
基因组学中的群体遗传结构分析与人类进化研究一、引言基因组学是研究生物个体遗传物质DNA的结构、功能和变异的科学,而群体遗传结构分析是基因组学研究的重要方向之一。
本文将重点探讨群体遗传结构分析在人类进化研究中的应用。
二、单倍型与单核苷酸多态性群体遗传结构分析主要依赖于单倍型和单核苷酸多态性的研究。
单倍型指的是一段DNA片段在群体中的存在形式,常用单倍型标记来描述个体间的遗传关系。
而单核苷酸多态性则是指个体间的遗传变异可以通过单个碱基的变化来衡量。
三、群体遗传结构分析方法1. 核苷酸多态性分析:通过测定群体中一定数量的SNP(单核苷酸多态性)位点,来推断个体之间的遗传关系和群体结构。
2. STR分析:短串联重复序列(STR)是一种多态性DNA标记,通过测定STR位点上的重复序列长度差异来鉴定个体之间的遗传关系。
3. 基于DNA指纹的分析:DNA指纹技术是通过测定个体DNA中特定的标记位点,如VNTR或SSR等来鉴定个体之间的遗传关系和群体结构。
4. 基因组重测序:随着高通量测序技术的发展,基因组重测序成为了研究群体遗传结构的重要手段。
四、群体遗传结构分析在人类进化研究中的应用1. 人类起源与迁移:通过分析不同地理区域的群体遗传结构差异,可以推测人类起源和迁移的历史。
例如,非洲原始人种的遗传多样性远远高于其他地区的现代人种,这与人类起源于非洲的观点相一致。
2. 自然选择与遗传适应:通过比较不同环境条件下的群体遗传结构,可以研究自然选择和遗传适应的作用。
例如,高海拔地区的人群相对于低海拔地区的人群通常会表现出一些形态和生理方面的适应性特征,这与缺氧环境对基因的选择性作用有关。
3. 疾病易感性:群体遗传结构分析可以帮助我们研究人群中疾病易感基因的分布规律,并提供指导针对性的疾病预防和治疗策略。
例如,通过分析不同人群中BRCA1和BRCA2等乳腺癌易感基因的突变频率,可以为乳腺癌的早期筛查和治疗提供依据。
五、群体遗传结构分析的挑战与前景1. 数据处理与分析的挑战:随着测序技术的迅速发展,群体遗传结构分析所产生的数据量也在不断增加,对数据处理与分析能力提出了更高的要求。
10.(2019年广西北部湾)(2分)下列有关生物生殖发育的叙述,正确的是()A.用马铃薯的块茎繁殖新植株属于有性生殖B.在适宜的条件下,鸟卵均能孵化成雏鸟C.青蛙的生殖发育过程离不开水,幼体和成体均用鳃呼吸D.蝗虫的发育过程经历了受精卵、若虫和成虫三个阶段12.((2019年江西省)1分)关于“稻花香里说丰年,听取蛙声一片“蕴含的生物学知识,下列叙述错误的是()A.水稻通过种子繁殖,属于有性生殖B.雄蛙鸣叫吸引雌蛙抱对,有助于通过体内受精繁殖后代C.青蛙捕食害虫有利于水稻丰收D.水稻、青蛙结构和功能的基本单位都是细胞1,(2019年甘肃省定西市)蜂鸟是世界上最小的鸟类,它的卵仅重1g,而鸵鸟卵可重达1500g。
无论蜂鸟卵还是鸵鸟卵,将来发育成雏鸟的结构都是()A. 卵白B. 卵黄C. 胚盘D. 卵黄系带13.(2019年河北省保定市)(1分)下列关于生物生殖和发育的叙述,正确的是()A.人的受精卵在输卵管内形成B.苍蝇的发育属于不完全变态C.组织培养和试管婴儿均属于无性生殖D.青蛙生殖发育特点是体内受精、变态发育6.(2019年黑龙江省龙东)(1分)人的受精卵形成的部位是()A.子宫B.输卵管C.卵巢D.阴道21.(2019年湖南省长沙市)(2分)下列动物的发育方式与如图所示动物一致的是()A.家蚕B.蜜蜂C.蝗虫D.苍蝇15.(2019年黑龙江省龙东)(1分)鸟的受精卵中能发育成雏鸟的结构是()A.卵黄B.卵白C.胚盘D.气室38. (2019贵州省六盘水市)下列关于生物生殖和发育的说法正确的是A.用嫁接或扦插的方法繁殖可保持果树的优良性状B.鸟卵的结构中,卵黄将来发育成雏鸟C.蝗虫的发育包括受精卵、幼虫、蛹和成虫四个时期D.雌青蛙产在水中的卵是受精卵19. (2019年湖北省黄冈市)生物通过生殖和发育,使得生命在生物圈中世代相续,生生不息。
下列有关叙述错误的是A. 卵巢是女件的主要生殖器官,女性月经的形成与卵巢分泌的激素有关B. 利用嫁接、扦插等无性生殖方式繁殖的新个体,只具有母体的遗传特性C. 鸟卵的卵黄和卵黄外面的卵內都能为胚胎发育提供营养D. 雌雄蛙抱对,完成体内受精,这有利于提高靑蛙的受精率7.(2019年江苏省盐城市)下列关于鸟类生殖与发育过程的叙述,正确的是()A.受精的鸡卵能在人工孵化箱中发育成雏鸡,主要原因是养料充足B.家鸽雏鸟的特点是腿足有力C.鸟类的受精方式为体内受精D.受精的鸟卵都在体外开始发育22.(2019年黑龙江省齐齐哈尔市)(2分)蝗虫与家蚕发育过程相比较,蝗虫不经过的时期是()A.成虫B.卵C.蛹D.幼虫34.(2019年海南省)下列关于动植物生殖、发育的说法正确的是()A.生殖期的青蛙雌雄抱对,体内受精,水中变态发育B.毛毛虫长大后变成美丽的蝴蝶要经过卵、幼虫、成虫期C.鸟的受精卵从母体产出后,在亲鸟的孵化下才开始发育D.用嫁接的方法可实现同一棵桃树上结出不同口味的桃子24.(2019年湖北省宜昌市)(2分)有关动植物生殖和发育的叙述,正确的是()A.“有心栽花花不开,无心插柳柳成荫”描述了柳树的有性生殖B.“菜青虫”在菜粉蝶一生中所处的时期是幼虫期C.青蛙和蝗虫都进行不完全变态发育D.求偶、交配、筑巢、产卵是所有鸟类共有的繁殖行为6.(2019年海南省)成熟的胎儿从母体产出的通道是图中的()A.① B.② C.③ D.④23.(2019年湖南省湘潭市)如图是鸟卵结构示意图。
诺禾致源最新“高性价比”群体进化研究成果
继2013年合作完成地山雀基因组测序之后,北京诺禾致源重测序事业部团队与中国科学院动物研究所研究人
员再次携手,通过对13个地区的32只大山雀进行全基因组重测序,解析了喜马拉雅山脉东部大山雀对随海拨
变化的气候的适应机制。
研究成果发表于2015年9月的Scientific Reports杂志(IF:5.578)。
其中,中国科
学院动物研究所屈延华研究员、诺禾致源田仕林为论文的共同第一作者。
群体重测序揭示
大山雀适应随海拨而变的气候的机制
NGS项目文章
研究背景
大山雀(Parus major )隶属于雀形目(Passeriformes)山雀科(Paridae)山雀属(Parus spilonotus )。
在东亚,主要分布在低海拔地区,也有部分种群生活在喜马拉雅山脉东部的高海拔地区。
有季节性迁徙习性,在海拔4000m地区繁殖,在海拔2000m地区越冬。
本研究采用群体重测序技术,从基因组水平上揭示了喜马拉雅山脉东部大山雀的起源及对这种季节性的、随海拨高度变化的气候的适应性机制。
研究方法
基于Illumina HiSeq 2000 测序平台,对来自13个地区的32只大山雀进行全基因组重测序,其中,11只大山雀来自喜马拉雅山脉东部地区,11只来自中国中/东部地区,10只来自内蒙古和蒙古,测序深度5X/样。
以近缘物种地山雀(Pseudopodoces humilis )基因组作为参考基因组,对大山雀群体进行了遗传多样性、种群历史动态、选择消除等分析。
研究结果
1. 大山雀的群体进化分析
群体遗传多样性分析表明,来自蒙古(MON)、东喜马拉雅山脉(EH)
和中国中/东地区(CE)的大山雀各自聚为一类。
EH和CE的亲缘关系较MON
更近。
大山雀和地山雀约在5.8-13.3百万年前发生了分化;0.7-2.8百万年前
大山雀中分化出了MON分支;0.4-1.9百万年前EH和CE发生了分化。
2. 大山雀种群历史动态分析
EH、CE和MON在0.3-0.4百万年前种群遭遇了瓶颈效应。
CE在0.06百万
年前有效群体大小迅速扩张,EH在同一时间开始扩张,但变化较平缓。
而
MON经历了一个漫长的瓶颈,一直持续到0.02百万年前,在末次盛冰期有效
群体大小稍有增加。
以上结果表明,EH种群动态受冰期气候影响小,这是由
于东喜马拉雅山脉局部环境相对稳定,长期生活在该地区的大山雀发生了高
海拔适应性进化。
3. 大山雀对高海拔的适应机制
通过选择消除分析,在东喜马拉雅山脉大山雀检测到183个基因受到强选
择,主要参与能量代谢过程和低氧反应。
(1)能量代谢基因进化:东喜马拉雅大山雀糖类代谢相关的基因发生了
快速进化,包括氨基糖和核苷酸糖代谢(5个基因)和胰岛素信号途径(11个
基因)。
大山雀这种适应机制适合其在中、高海拔季节性迁徙的生活习性。
(2)低氧适应:低氧反应基因富集在MAPK信号通路,调节东喜马拉雅
大山雀体内相关基因的表达来适应低氧环境。
(3)形态进化:东喜马拉雅大山雀骨骼发育相关的基因发生了快速进
化,与分布在中国中/东低地区域的大山雀相比,体型更大,有利于保存热
量。
此外,体型大倾向于有较高的氧亲和力,这也是东喜马拉雅大山雀能够
更好地调节体温适应高海拔的寒冷气候的一个原因。
参考文献
Qu YH, Tian SL, et al. Genetic responses to seasonal variation in altitudinal stress: whole-genome resequencing of great tit in eastern Himalayas. Scientific Reports, 2015. 图1 大山雀遗传多样性及分化时间 图2
调控低氧反应的受选择基因。