Leigh综合症
- 格式:ppt
- 大小:12.05 MB
- 文档页数:31


雷诺综合征(雷诺现象)
雷诺综合征,又称雷诺现象,是一种罕见疾病,表现为手指或脚趾在寒冷或情绪激动等情况下变得发紫、冰冷及疼痛,主要是由于血管对寒冷的异常反应所致。
本文将介绍雷诺综合征的症状、病因、诊断以及治疗方法。
症状
雷诺综合征的主要症状包括:
1.手指或脚趾发紫、冰冷
2.疼痛或刺痛感
3.感觉异常
4.容易出现手脚的皮肤溃疡
这些症状多在寒冷或情绪激动时出现,通常对称性发作。
病因
雷诺综合征的病因目前尚不十分清楚,可能与以下因素有关:
1.血管异常:雷诺综合征患者的血管对寒冷刺激存在异常反应,导致血
管痉挛和微循环障碍。
2.神经因素:神经系统的异常活动可能在雷诺综合征的发病机制中起着
重要作用。
诊断
雷诺综合征的诊断通常基于患者的症状和体征,医生可能还会进行以下检查:
1.冷试验:让患者将手指或脚趾浸入冷水中,观察反应。
2.血液检查:检查患者的血液循环情况。
3.血管造影:通过影像学检查来观察血管情况。
治疗
治疗雷诺综合征的方法包括:
1.避免寒冷刺激:尽量避免寒冷天气或佩戴保暖的手套、袜子等。
2.药物治疗:如钙通道阻滞剂、血管扩张药等。
3.物理治疗:如温热疗法可以改善患者的血液循环。
4.手术治疗:对于严重的雷诺综合征患者,可能需要进行手术治疗。
总之,雷诺综合征是一种影响生活质量的疾病,但随着医疗技术的不断进步,患者可以通过综合治疗得到有效控制。
及时就医并积极治疗是关键。



mt-atp6基因
MT-ATP6基因是线粒体DNA中的一种基因,它提供了制造功能蛋白所必需的指令。
这种基因的突变可以导致一系列的疾病,其中之一就是Leigh综合征。
Leigh综合征是一种罕见的神经系统遗传性疾病,主要表现为神经系统的发育障碍和进行性的脑损伤。
这种疾病通常在婴儿期或幼儿期发病,其临床表现复杂多样,起病隐匿,早期不易识别。
MT-ATP6基因的突变可以改变ATP合酶的结构或功能,降低线粒体产生ATP的能力。
当这个变异出现在一个人的更高比例的时候,其线粒体大于90%至95%,导致更严重的病状,称为母系遗传Leigh综合征。
对于Leigh综合征,目前尚无特异治疗方案,临床以对症处理为主,补充线粒体氧化呼吸链中的相关辅酶,部分患者在避免感染等应激因素后,病情可缓慢发展,甚至能维持长期稳定。
同时,对于这类疾病,高危儿随访中遇到精神运动发育落后的患儿时,应全面分析病因,避免遗漏询问家族遗传病史,加强多学科协作,早期诊断与早期治疗,减轻患儿病痛及家庭负担。

rieger 综合症,属常染色体显性遗传病,多为双侧性。
男女患病机会均等。
本病角膜、虹膜和前房的异常是眼前部中胚叶发育不良的一组遗传性疾病。
有人认为胚胎于 .J6?I 时发育障碍有关。
角膜及前房的异常主要表现为:角膜缘的界线常难以分辨,大多数患者大角膜,少数为小角膜L’M。
在角膜内缘呈玻璃样半透明环,通常称为后胚胎环,它是接近房角处的角膜中胚叶组织的增殖。
房角镜下可见周边虹膜有大的条索可遮盖部分或全部小梁,是继发青光眼的主要原因L)M。
虹膜异常,常表现在虹膜基质层发育不良。
瞳孔异常主要表现瞳孔缘色素层外翻,虹膜裂孔,瞳孔移位等。
"#$%$& 异常伴有面骨的发育畸形和牙齿的发育异常时,常称为"#$%$& 综合症L’:)M。
"#$%$& 综合症的治疗与开角型青光眼相似,主要是控制青光眼应及早行抗青光眼手术,本病例双眼继发性青光眼,经药物治Axenfeld异常的患者并不十分罕见,但遗憾的是,由于日常工作的繁忙。
一些医师可能会对可疑的患者漏诊。
通过复习该病的一些相关文献,及一些病例观察。
认为应当将Axenfeld异常归纳入Axenfeld-rieger症候群。
并且,建议1、认真进行角膜缘和房角检查2、谨慎看待UBM结果。
避免错误地测量房角3、对可疑的患者行基因检查,预测家族中子代的发病几率。
讨论:1、患者此前曾多次眼科检查。
但由于患者的特殊不典型性:角膜后胚环不明显;房角入射角部分类似高原虹膜形态;虹膜与Schwalbe线粘连疏松;眼压多次测量正常范围。
以上特点导致患者长期未得到确诊。
应当引起重视。
2、关于先天性房角异常。
先天性房角异常源于细胞分化异常或胚胎期组织退化滞后。
其最常见的良性改变为角膜后胚胎环。
正常的Schwalbe线为小梁网和角膜后弹力层终点的连接处。
角膜后胚胎环表现为Schwalbe线加重、突起和向中央移位,裂隙灯下显示白色、明亮的不规则弧线,距角膜缘0.5~2.0mm。


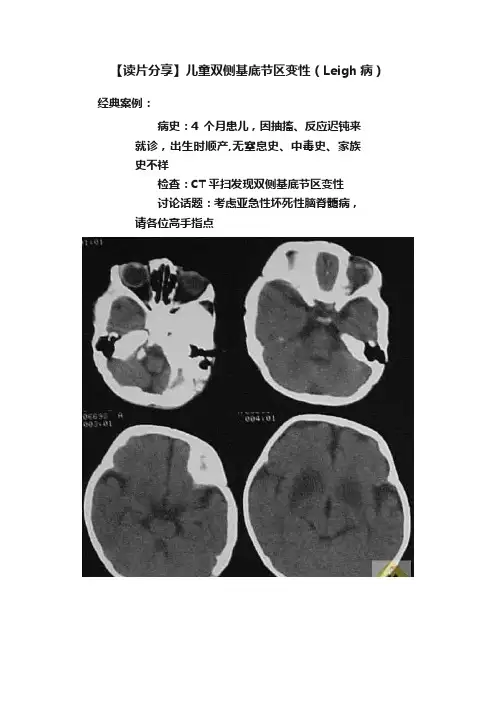
【读片分享】儿童双侧基底节区变性(Leigh病)经典案例:病史:4个月患儿,因抽搐、反应迟钝来就诊,出生时顺产,无窒息史、中毒史、家族史不祥检查:CT平扫发现双侧基底节区变性讨论话题:考虑亚急性坏死性脑脊髓病,请各位高手指点案例讨论:发言1:支持亚急性坏死性脑病分析:本病又称Leigh氏综合征,病因不明,可能与硫胺有关的一种先天性代谢障碍。
乳儿期缓慢起病,有家族史。
进行性视,听力及智力障碍。
共济失调,肌力及肌张力低下,一般发病后2-3年因球麻痹而出现吞咽和呼吸困难而死亡。
CT典型表现为两侧壳核对称性低密度区,无增强。
发言2:支持Leigh氏综合征,肝豆状核变性年龄太小基底节病变:1、肝豆状核变性;2、亚急性坏死性脑病;3、Kearns-Sayre综合征;4、弥漫性躯体毛细血管扩张疣;5、苍白球黑质色素变性;6、一氧化碳中毒;7、其他中毒;8、病毒性脑炎;9、脑血管病;10、维生素B1缺乏症。
亚急性坏死性脑脊髓病又称Leigh综合征,是一种罕见的常染色体隐性遗传性神经系统变性的疾病,主要累及婴幼儿,与硫胺代谢先天性紊乱有关。
此病于1951年由Leigh首先报道,故又称Leigh综合征。
其主要病理变化为双侧壳核、尾状核、苍白球坏死,小脑、脑干、延髓、脊髓均可累及。
临床表现有:1、不明原因的营养不良及肝、胃肠疾病症状。
2、进行性智力衰退及听力障碍。
3、开始可有肢体无力及视力减退,眼球运动障碍、眼震、抽搐等。
4、病情恶化后呈木僵状态,肌强直阵挛,可出现球麻痹及呼吸困难而死亡。
实验室检查无特异性发现,血乳酸丙酮酸增高,血白细胞增高,脑脊液检查蛋白轻度增加。
脑CT基底节、丘脑、脑干出现低密度影,大脑皮层可出现萎缩。
本病目前尚无特殊的治疗方法,预后不良,多于发病后数月至数年死亡。
发言3:支持亚急性坏死性脑脊髓病CT表现为双侧基底节对称性低密度病变种类颇多,根据病因可分为:代谢性疾病、感染性疾病、中毒性疾病、脑血管性及缺氧性疾病。
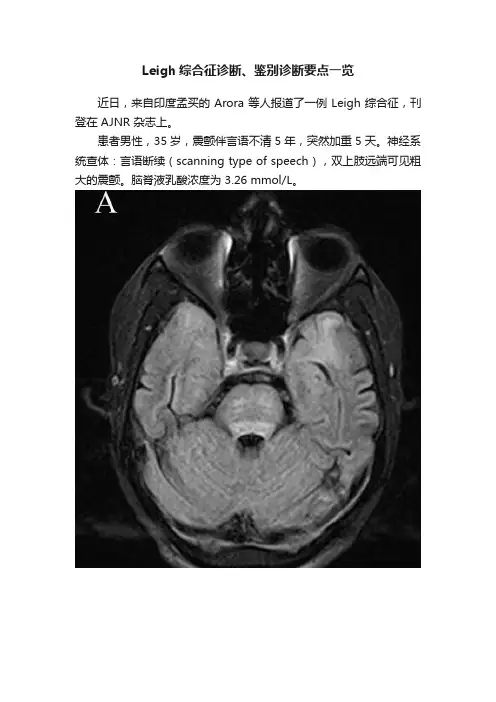
Leigh综合征诊断、鉴别诊断要点一览近日,来自印度孟买的 Arora 等人报道了一例 Leigh 综合征,刊登在 AJNR 杂志上。
患者男性,35 岁,震颤伴言语不清 5 年,突然加重 5 天。
神经系统查体:言语断续(scanning type of speech),双上肢远端可见粗大的震颤。
脑脊液乳酸浓度为 3.26 mmol/L。
图 A~D 头颅 MRI FLAIR 序列显示延髓、脑桥、中脑、丘脑、壳核、尾状核和双侧额叶、顶叶、侧脑室周围白质及两侧外囊均呈高信号图 E~F 短 TE(图 E)和中间 TE(图 F)MR 波谱成像显示基线伪迹(baseline artifact),但仍可见升高的乳酸水平——1.3ppm最终诊断:Leigh 综合征Leigh 综合征,又称亚急性坏死性脑脊髓病(SNEM),是一种罕见的、累及中枢神经系统的线粒体疾病;线粒体 DNA 突变或核 DNA 突变(SURF-1、COX 基因)均可引起氧化磷酸化(ATP 的产物)的损害,这已经被证实为Leigh 综合征的病因。
长期能源不足导致神经元丢失和神经胶质增生,最常累及基底节区和脑干。
临床特点由于成年型Leigh 综合征的病例少有报道,故临床特征总结尚不完善。
该病通常发生在婴儿期,常见的表现有腹泻、呕吐、发育迟滞、肌张力低,通常在几年内死亡;成年型Leigh 综合征的临床表现有肌张力低、共济失调、眼肌麻痹和震颤、精神异常、自主神经功能障碍、睡眠障碍,以及癫痫发作。
该病起病隐袭,呈间歇性进展,晚期呈亚急性或急性恶化;呼吸衰竭是该病最常见的死亡原因。
心血管症状通常由肥厚性心肌病或非对称性心室间隔肥厚引起,该病还可累及周围神经系统。
诊断要点MRI T2 像和 FLAIR 序列上,脑干、导水管周围灰质、壳核、尾状核头和丘脑呈高信号;MRI T1 像上,上述区域则信号减低。
在急性期,上述区域可出现扩散受限;MR 波谱成像可见乳酸峰值为 1.3ppm。

Leigh综合征一例病例报告1广东省东莞市常平医院神经内科广东东莞 523560;2中山大学附属第一医院神经科广东广州 510080Leigh综合征(Leigh Syndrome,LS)是一种少见病,又称为亚急性坏死性脑脊髓病(Subacute Necrotizing Encephalomyelopathy,SNE)。
LS是一种线粒体脑肌病,它与线粒体酶系统代谢异常有关。
该病绝大多数起病于婴幼儿,逐渐进展,成年人发病较为罕见。
我们在2014-07-28收治了一例成年患者,临床诊断为亚急性坏死性脑脊髓病,且合并腓骨肌萎缩症2型,特报道如下。
1病例资料1.1 病史患者,男性,29岁,广东省兴宁市某单位职员,主因“发热,四肢乏力20余天,加重4天”于2014年07月28日入院。
患者6月30日无明显诱因出现发热,感四肢乏力,无感冒咳嗽等症状,体温最高达40摄氏度,无寒战,至广州市第一人民医院就诊(具体情况不详)。
7月1日热退,仍有乏力,但生活可自理,间断有发热,最高达38.5摄氏度,轻微头痛,排尿费力,左手指有麻木。
无头晕,恶心,呕吐,肌肉疼痛。
7月24日四肢乏力加重,卧床不起,精神差,送至我院急诊。
查头部CT示双侧丘脑低密度病灶,予以营养神经,营养支持,头孢呋辛抗感染等对症支持治疗,7月25日出现解小便不出,予以插尿管。
其后双下肢乏力进一步加重,思睡,较难唤醒,拟“四肢乏力查因”收住我病区。
起病以来,患者精神极差,无抽搐,有大便失禁,食欲欠佳。
近来体重下降情况不详。
既往高中起就有双下肢肌肉明显萎缩,2013年在我院门诊诊断为腓骨肌萎缩症。
1.2 入院查体T:38℃ P:100次/分 R:19次/分 BP:122/69mmHg。
查体欠合作。
心肺腹检查未见异常。
专科体查:嗜睡,表情淡漠,情绪低落,语缓,对答仅回答简单字词,时间,地点,人物定向正确。
无法讲出鸡与鸭的不同。
远近记忆力正常,计算力正确,但对答缓慢。
Leigh综合征临床表现及其基因报告1例周红亮【期刊名称】《中国中西医结合儿科学》【年(卷),期】2017(009)005【总页数】2页(P459-460)【关键词】Leigh综合征;基因检测;儿童【作者】周红亮【作者单位】222000江苏连云港,连云港市第二人民医院儿科【正文语种】中文【中图分类】R725Leigh综合征又称亚急性坏死性脑脊髓病,是线粒体呼吸链缺陷导致机体能量代谢障碍,以心、脑和骨骼肌等能量需求大的组织器官损害为著,发病率约为活产儿的1/40 000。
我国Leigh综合征临床与基因共同诊断病例文献较少,本文分析1例Leigh综合征患儿的临床表现、辅助检查与基因型特点,现报道如下。
患儿,女,1岁1个月,2016年3月2日初诊。
因“四肢无力1周伴纳差”入院。
查体:体温37 ℃,脉搏145次/分,身高80 cm,体质量8.5 kg,血压61/36 mm Hg。
神志清,反应欠佳,无脱水貌,双侧瞳孔等圆等大,对光反射灵敏,口角无歪斜,颈部无抵抗,前囟张力正常,无气促,双肺呼吸音粗,无啰音,心音有力,腹平软,肝、脾未扪及肿大,肠鸣音正常。
肌力左上肢3级,右上肢3级,左下肢2级,右下肢2级,肌张力低下,四肢末梢循环可。
辅助检查:生化检验:血氨23 μmol/L(正常),乳酸3.2 mmol/L(升高),脑脊液基本正常,TORCH均阴性,血尿气相质谱未见异常,细胞免疫及体液免疫正常,头颅MRI:脑功能成像(DWI)3.0,两侧基底节区、两侧大脑脚异常信号影,考虑代谢性疾病可能。
肝肾功能电解质正常。
诊断:肌无力原因待查,遗传性代谢病;入院后完善相关检查助诊。
入院完善基因等相关检查,予营养神经,补液及对症治疗,病情稳定后出院等待基因检测结果。
2016年6月13日结合病史及基因检测结果,诊断Leigh综合征,其基因报告分别见表1、2。
表2报告中的“样本中突变率”仅限于在本次检测样本组织(如外周血白细胞)中的突变率,并不等同于受检者的其他组织的突变率。
Leigh综合征及两种肿瘤的分子遗传学研究的开题报
告
题目:Leigh综合征及两种肿瘤的分子遗传学研究
背景及研究意义
Leigh综合征是一种罕见的神经变性疾病,通常在幼儿期出现,主要表现为进行性双侧锥体外系损害、脑干病变和肌无力,导致智力残疾和生命威胁。
两种肿瘤分别为乳腺癌和结直肠癌,均是常见恶性肿瘤。
癌症是一种复杂的多基因疾病,其发病与遗传因素密切相关。
了解乳腺癌和结直肠癌发病的分子遗传学机制,对于其诊断、治疗以及基因治疗的开发具有重要意义。
因此,本研究旨在通过分子遗传学研究,探讨Leigh综合征和两种恶性肿瘤的遗传学机制,为相关疾病的早期诊断和治疗提供理论依据。
研究方案
1. 研究对象
本研究将选取患有Leigh综合征或乳腺癌/结直肠癌的患者,以及健康人作为对照组。
2. 样本采集
收集患者和对照组的血液或组织样本,提取DNA进行基因测序,获取个体基因组数据。
3. 数据分析
利用生物信息学方法对基因组数据进行分析,寻找Leigh综合征和乳腺癌/结直肠癌的遗传相关基因。
结合临床资料进行进一步分析,探讨患者遗传变异与疾病发生的关系,确定其相关性。
4. 西方博士
通过Western Blot方法检测标志性蛋白的表达量,探讨遗传变异对蛋白表达量的影响,进一步验证遗传变异和疾病的关系。
预期结果及意义
本研究将探讨Leigh综合征和乳腺癌/结直肠癌的分子遗传学机制,发现与疾病发生相关的基因或遗传变异,为相关疾病的早期诊断和治疗提供依据,并为基因治疗的开发提供参考。
同时,本研究还有望拓展基因治疗的范围,为人们解决基因遗传性疾病带来希望。