原发性血小板增多症的诊断与治疗文稿演示
- 格式:ppt
- 大小:8.73 MB
- 文档页数:25



原发性血小板增多症(ET)的诊疗1:骨髓细胞:骨髓为主要造血组织,产生红细胞、粒细胞、单核细胞、淋巴细胞和血小板等,故骨髓细胞包括各种血细胞系的不同发育阶段的细胞。
如粒细胞系:原粒细胞/早幼粒细胞/中幼粒细胞/晚幼粒细胞/杆状粒细胞/分叶核粒细胞;淋巴细胞系:原淋巴细胞/幼淋巴细胞/淋巴细胞;红细胞系:原红细胞/早幼红细胞/中幼红细胞/晚幼红细胞/网织红细胞/红细胞;单核细胞系:原单核细胞/幼单核细胞/单核细胞;巨核细胞系:原巨核细胞/幼巨核细胞/巨核细胞/最后形成血小板;浆细胞系:亦称效应B细胞,免疫系统中释放大量抗体的细胞,包括原浆细胞、幼浆细胞和浆细胞。
还含有其它细胞,如网状细胞、内皮细胞(吞噬细胞)等。
某些化学物质(如苯)抑制骨髓细胞分裂增殖能力,造成白细胞减少、血小板减少、再生障碍性贫血,或刺激粒细胞系过度增生,诱发白血病。
2:骨髓增殖性肿瘤(MPN)(也称慢性骨髓增殖性疾病):指分化相对成熟的一系或多系骨髓细胞持续克隆性增殖所致的一组造血系统肿瘤性疾病。
表现为一种或多种血细胞的质和量异常,伴肝、脾或淋巴结肿大。
病因及发病机制尚不完全明确,目前认为Janus 型酪氨酸激酶2(JAK2)基因突变致酪氨酸激酶信号途径(JAK-STAT)过度活化与该组病发病密切相关。
MPN分8类:慢性髓性白血病(又称慢性粒细胞白血病)、慢性中性粒细胞白血病、慢性嗜酸性粒细胞白血病/高嗜酸性粒细胞综合征、真性红细胞增多症、原发性血小板增多症、原发性骨髓纤维化、肥大细胞增多症和不能分类 MPN。
常见4类:慢性髓性白血病、真性红细胞增多症、原发性血小板增多症、原发性骨髓纤维化。
本组病共同特征:①病变发生在多能造血干细胞(为骨髓中原始造血干细胞/具有自我更新和分化为各种谱系造血细胞的能力)。
②各病以骨髓某系细胞恶性增殖为主,同时均有不同程度累及其他系造血细胞的表现。
③细胞增生还可发生于脾、肝、淋巴结等髓外组织,即髓外造血髓外造血(指在疾病或骨髓代偿功能不足时,肝、脾、淋巴结可恢复胚胎时期的造血功能)。




2023原发性血小板增多症诊断和治疗原发性血小板增多症(ET)是费城染色体阴性的慢性骨髓增殖性肿瘤(MPN)中较常见的亚型,年发病率为1~2.5∕10万,发病高峰年龄在50-70岁。
ET 起源于骨髓造血干/祖细胞的克隆性疾病,表现为巨核细胞过度增殖从而导致血小板计数明显增高。
ET的发病机制为基因突变或其他因素导致JAK-STAT信号通路高度活化。
ET的驱动基因突变包括JAK2V617F、钙网蛋白基因(CA1R)及骨髓增殖性白血病蛋白基因(MP1)突变,分另!!占50%~60%、15%~35%及2%~4%.20%~30%的患者存在MPN非特异性基因突变(包括信号通路、转录因子、DNA甲基化、组蛋白甲基化以及剪接基因等\一、典型病例患者,女,28岁,因孕检发现”血小板增多4周”入院。
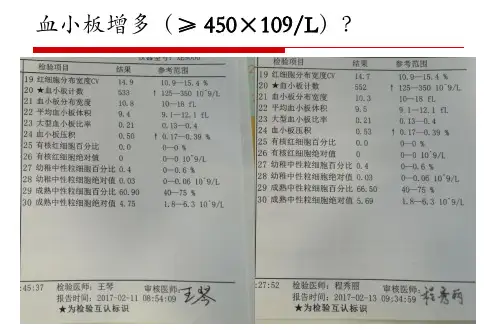
入院4周前血常规:WBC1o.53χ109/1、RBC4.6×1012∕1x HGB123g∕1x红细胞压积(HCT)36%、P1T1180×109/1、中性粒细胞绝对计数ANC)7.4×109/1,无不适症状。
3d前复查血常规:WBC9.46χ109/1、RBC4.8×1012∕1x HGB130g/1、HCT38%、P1T1460×109∕1s ANC6.9×109∕1z未治疗。
既往体健,无吸烟、饮酒史,妊娠16周,孕1产0,否认家族类似病史及遗传病史。
入院后血常规:WBC9.56χ109/1、RBC4.7×1012∕1x HGB125g/1、HCT37%、平均红细胞体积(MCV)85.7f1、平均红细胞血红蛋白含量(MCH)28pg、平均红细胞血红蛋白浓度(MCHC)327g/1、P1T1582×109∕1.ANC7.1×109∕1;外周血乳酸脱氢酶(1DH)176U∕1(参考值0~248U/1);血管性血友病因子(VWF)抗原77%(参考值50%~160%),VWF活性58.1%(参考值48.8%~163.4%);肝肾功能、电解质、铁代谢、C反应蛋白、凝血功能、肿瘤标志物、抗核抗体谱、狼疮抗凝物及抗磷脂抗体均未见异常。


原发性血小板增多症1例【关键词】原发性血小板增多;出血倾向;血栓形成【中图分类号】r725.5 【文献标识码】a 【文章编号】1004-7484(2012)06-0477-01原发性血小板增多症(primary throbocythemia)是骨髓增生性疾病,其特征为出血倾向及血栓形成,外周血血小板持续明显增多,功能也不正常,骨髓巨核细胞过度增殖。
由于本病常有反复出血,故也名为出血性血小板增多症,发病率不高,多见40岁以上者。
1 临床资料1.1 患者男 85岁主因慢性咳嗽、咳痰、喘息6年,复发10天入院。
入院前6年每于感冒后出现咳嗽、咳痰,天气转暖或经治疗后好转,此后每年冬季或受凉后发作,发作持续2-4个月不等,经治疗好转,此后间断复发,缓解期生活能自理,但病情逐年加重;10天前因感冒后病情复发入院,住院期间出现右侧肩胛部血肿,经治疗后好转。
出院1个月后因右侧肩背部血肿再次入院治疗。
1.2 查体:t37.0℃ p88次/分 r22次/分 bp190/90mmhg 神清,精神欠佳,轻度喘息貌,口唇无紫绀,双侧巩膜无黄染,结膜无水肿,双肺呼吸音粗,双肺可闻及散在哮鸣音,左肺为重,双肺底可闻及少许湿啰音,心率108次/分,节律绝对不齐,第一心音强弱不等,各瓣膜听诊区未闻及病理性杂音,腹平软,未见胃肠型及蠕动波,无反跳痛及肌紧张,肝脾肋下未触及,叩诊呈鼔音,移动性浊音阴性,肠鸣音3次/分,双下肢无水肿,四肢肌力及肌张力正常,病理反射均阴性。
1.3 辅助检查:入院时胸片报告为慢性支气管炎合并感染;b超结果:左右房扩大,室间隔基底部运动幅度减低,主动脉瓣反射增强,各瓣膜轻度返流,轻度肺动脉高压,左心功能减低,肝实质弥漫性病变,胆囊壁不光滑,脾大,左肾囊肿;血电解质、心肌酶、尿常规、肝功能、血糖、肾功能正常,心电图示异位心律-心房纤颤,心室率108次/分;2010-12-15血常规:wbc22.9*109/l,plt630*109/l,pct0.50%,嗜中性粒细胞百分比88.3%,巨大不成熟细胞百分比2.9%,嗜酸性粒细胞1.15*109/l,bas:0.21*109/l,巨大不成熟细胞0.64*109/l;2011-1-24血常规:wbc26.6*109/l,plt723*109/l,pct0.546%,嗜中性粒细胞百分比84.2%,巨大不成熟细胞百分比2.2%,嗜酸性粒细胞1.14*109/l,bas:0.35*109/l,巨大不成熟细胞0.57*109/l;1.4 既往否认肝炎病史,无长期饮酒史。
原发性血小板增多症的鉴别诊断
*导读:原发性血小板增多症(primarythrobocythemia)是骨髓增生性疾病,其特征为出血倾向及血栓形成,外周血血小板持续明显增多,功能也不正常,骨髓巨核细胞过度增殖。
由于本病常有反复出血,故也名为出血性血小板增多症,发病率不高,多见40岁以上者。
……
原发性血小板增多症(primary throbocythemia)是骨髓增生性疾病,其特征为出血倾向及血栓形成,外周血血小板持续明显增多,功能也不正常,骨髓巨核细胞过度增殖。
由于本病常有反复出血,故也名为出血性血小板增多症,发病率不高,多见40岁以上者。
鉴别诊断:
1.继发性血小板增多症:可见于生理性和病理性两大类。
生理性见于运动后和分娩时或注射肾上腺素后。
病理性可见于各种急、慢性感染,慢性失血后,恶性肿瘤,外伤手术、脾切除后,结缔组织病,结核,肾上腺机能亢进等。
其特点为血小板计数小于1000×109/L,少见出血及微血管栓塞表现,脾脏一般不肿大,同时在短期内即恢复。
2.其他骨髓增殖性疾病:主要应与慢性粒细胞白血病、真性红细胞增多症、骨髓纤维化等加以鉴别。
慢性粒细胞白血病以外周血及骨髓中见到各阶段幼稚粒细胞及嗜碱性粒细胞增多为主,可见
ph1染色体,脾大明显;真性红细胞增多症以红系细胞增多较为明显,血红蛋白增多,男性180g/L,女性170g/L;骨髓纤维化则是骨髓发生弥漫性纤维组织和骨髓增生伴髓外造血的一种骨髓增生性疾病,主要表现为脾肿大及贫血。