2010年版《药典》微生物限度标准
- 格式:doc
- 大小:26.50 KB
- 文档页数:2

中国药典2010 年版一部附录附录Ⅰ A 丸剂丸剂系指饮片细粉或提取物加适宜的黏合剂或其他辅料制成的球形或类球形制剂,分为蜜丸、水蜜丸、水丸、糊丸、蜡丸和浓缩丸等类型。
蜜丸系指饮片细粉以蜂蜜为黏合剂制成的丸剂。
其中每丸重量在0.5g(含0.5g)以上的称大蜜丸,每丸重量在0.5g 以下的称小蜜丸。
水蜜丸系指饮片细粉以蜂蜜和水为黏合剂制成的丸剂。
水丸系指饮片细粉以水(或根据制法用黄酒、醋、稀药汁、糖液等)为黏合剂制成的丸剂。
糊丸系指饮片细粉以米粉、米糊或面糊等为黏合剂制成的丸剂。
蜡丸系指饮片细粉以蜂蜡为黏合剂制成的丸剂。
浓缩丸系指饮片或部分饮片提取浓缩后,与适宜的辅料或其余饮片细粉,以水、蜂蜜或蜂蜜和水为勤合剂制成的丸剂。
根据所用黏合剂的不同,分为浓缩水丸、浓缩蜜丸和浓缩水蜜丸。
丸剂在生产与贮藏期间应符合下列有关规定。
一、除另有规定外,供制丸剂用的药粉应为细粉或最细粉。
二、蜜丸所用蜂蜜须经炼制后使用,按炼蜜程度分为嫩蜜、中蜜和老蜜,制备蜜丸时可根据品种、气像等具体情况选用。
除另有规定外,用塑制法制备蜜丸时,炼蜜应雄热加入药粉中,混合均匀;处方中有树脂类、胶类及含挥发性成分的药味时,炼蜜应在60℃左右加入;用泛制法制备水蜜丸时,炼蜜应用沸水稀释后使用。
三、浓缩丸所用提取物应按制法规定,采用一定的方法提取浓缩制成。
四、除另有规定外,水蜜丸、水丸、浓缩水蜜丸和浓缩水丸均应在80℃以下干燥;含挥发性成分或淀粉较多的丸剂(包括糊丸)应在60℃以下干燥;不宜加热干燥的应采用其他适宜的方法干燥。
五、制备蜡丸所用的蜂蜡应符合本版药典该饮片项下的规定。
制备时,将蜂蜡加热熔化,待冷却至60℃左右按比例加入药粉,棍合均匀,趁热按塑制法制丸,并注意保温。
六、凡需包衣和打光的丸剂,应使用各品种制法项下规定的包衣材料进行包衣和打光。
七、丸剂外观应圆整均匀、色泽一致。
蜜丸应细腻滋润,软硬适中。
蜡丸表面应光滑无裂纹,丸内不得有蜡点和颗粒。


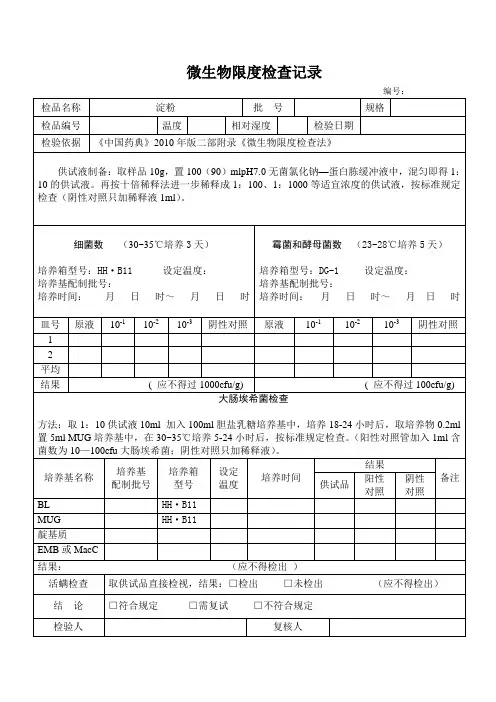
微生物限度检查标准操作规程⏹ 1. 目的:建立微生物限度检查的基本操作,为微生物检查人员提供正确的操作规程。
⏹ 2. 依据:《中华人民共和国药典》2010年版二部。
中华人民共和国卫生部《药品卫生检验方法》⏹ 3. 范围:本标准适用于QC化验室的微生物限度检查。
⏹ 4. 职责:QC微生物检验员对本标准的实施负责。
⏹ 5. 程序:⏹ 5.1. 定义:微生物限度检查法:微生物限度检查法系检查非规定灭菌制剂及其原料、辅料受微生物污染程度的方法。
⏹ 5.2. 实验用具:⏹电热恒温培养箱、电热恒温水温箱、试管、刻度吸管、量筒、三角瓶、培养皿、试管架、注射器、针头、注射器盒、研钵、75%酒精棉球、紫外灯(365nm波长)。
⏹ 5.3. 培养基:⏹ 5.3.1. 营养琼脂培养基、玫瑰红钠琼脂培养基、胆盐乳糖培养基、麦康凯琼脂培养基、枸橼酸盐培养基、磷酸盐葡萄糖胨水培养基、蛋白胨水培养基、4-甲基伞形酮葡萄糖苷酸(MUG)培养基。
⏹ 5.3.2. 培养基的管理、配置应符合检定菌、培养基管理规程、培养基配置规程。
⏹ 5.4. 试液:甲基红试液、а-奈酚乙醇试液、40%氢氧化钾溶液、靛基质试液。
⏹ 5.5. 稀释剂:0.9%无菌氯化钠溶液、PH7.0无菌氯化钠-蛋白胨缓冲液。
⏹ 5.6. 供试品溶液的制备:取供试品(供试品如为固体,置研钵中研磨成细粉)放入试管中,加入适量的稀释剂制成1:10浓度的供试品溶液。
⏹ 5.7. 对照用菌液:控制菌检查均应作相应已知菌的对照试验,对照菌株为大肠杆菌[CMCC(B)44102]。
⏹ 5.8. 检查法:⏹除另有规定外,细菌的培养温度为30-35℃,霉菌的培养温度为23-28℃,控制菌的培养温度为35-37℃。
⏹ 5.8.1. 细菌、霉菌计数:⏹ 5.8.1.1. 平皿法⏹采用平皿法进行菌数测定时,应取适宜的连续2~3个稀释级的供试液。
⏹取供试液1ml,置直径90mm的无菌平皿中,注入15~20ml温度不超过45℃的溶化的营养琼脂培养基或玫瑰红钠琼脂培养基或酵母浸出粉胨葡萄糖琼脂培养基,混匀,凝固,倒置培养。

化学药品质量控制按剂型分ⅠA片剂ⅠB注射剂ⅠC酊剂ⅠD栓剂ⅠE胶囊剂ⅠF软膏剂乳膏剂糊剂ⅠG眼用制剂ⅠH丸剂ⅠJ植入剂(增订)ⅠK糖浆剂ⅠL气雾剂粉雾剂喷雾剂ⅠM膜剂ⅠN颗粒剂ⅠO口服溶液剂口服混悬剂口服乳剂ⅠP散剂ⅠQ耳用制剂ⅠR鼻用制剂ⅠS洗剂冲洗剂灌肠剂ⅠT搽剂涂剂涂膜剂ⅠU凝胶剂ⅠV贴剂片剂系指药物与适宜的辅料混匀压制而成的圆片状或异形片状的固体制剂。
片剂以口服普通片为主,另有含片、舌下片、口腔贴片、咀嚼片、分散片、可溶片、泡腾片、阴道片、阴道泡腾片、缓释片、控释片与肠溶片等。
含片系指含于口腔中,药物缓慢溶解产生持久局部作用的片剂。
含片中的药物应是易溶性的,主要起局部消炎、杀菌、收敛、止痛或局部麻醉作用。
含片应进行释放度检查。
舌下片系指置于舌下能迅速溶化,药物经舌下黏膜吸收发挥全身作用的片剂。
舌下片中的药物与辅料应是易溶性的,主要适用于急症的治疗。
口腔贴片系指粘贴于口腔,经黏膜吸收后起局部或全身作用的片剂。
口腔贴片应进行溶出度或释放度检查。
咀嚼片系指于口腔中咀嚼或吮服使片剂溶化后吞服,在胃肠道中发挥作用或经胃肠道吸收发挥全身作用的片剂。
咀嚼片口感、外观均应良好,一般应选择甘露醇、山梨醇、蔗糖等水溶性辅料作填充剂和黏合剂。
咀嚼片的硬度应适宜。
分散片系指在水中能迅速崩解并均匀分散的片剂。
分散片中的药物应是难溶性的。
分散片可加水分散后口服,也可将分散片含于口中吮服或吞服。
分散片应进行溶出度检查。
可溶片系指临用前能溶解于水的非包衣片或薄膜包衣片剂。
可溶片应溶解于水中,溶液可呈轻微乳光。
可供外用、含漱等用。
泡腾片系指含有碳酸氢钠和有机酸,遇水可产生气体而呈泡腾状的片剂。
泡腾片中的药物应是易溶性的,加水产生气泡后应能溶解。
有机酸一般用枸橼酸、酒石酸、富马酸等。
阴道片与阴道泡腾片系指置于阴道内应用的片剂。
阴道片和阴道泡腾片的形状应易置于阴道内,可借助器具将阴道片送入阴道。
阴道片为普通片,在阴道内应易融化、崩解并释放药物,主要起局部消炎杀菌作用,也可给予性激素类药物。

《中国药典》2010年版二部附录XI J (附录115页)《微生物限度检查法》微生物限度标准非无菌药品的微生物限度标准是基于药品的给药途径及对患者健康潜在的危害而制订的。
药品的生产、贮存、销售过程中的检验,原料及辅料的检验,新药标准制订,进口药品标准复核,考察药品质量及仲裁等,除另有规定外,其微生物限度均以本标准为依据。
1.制剂通则、品种项下要求无菌的制剂及标示无菌的制剂应符合无菌检查法规定。
2.口服给药制剂细菌数每1g不得过l000CFU 。
每lml 不得过100CFU 。
霉菌和酵母菌数每lg或lml 不得过100CFU 。
大肠埃希菌每1g 或lml不得检出.3 .局部给药制剂3.1用于手术、烧伤及严重创伤的局部给药制剂应符合无菌检查法规定。
3.2 耳、鼻及呼吸道吸入给药制剂细菌数每1g、lml 或l0cm2,不得过100CPU 。
霉菌和酵母菌数每1g、lml 或l0cm2,不得过10CPU 。
金黄色葡萄球菌、铜绿假单胞菌每1g、lml 或l0cm2不得检出。
大肠埃希菌鼻及呼吸道给药的制剂,每1g、lml 或l0cm2,不得检出。
3.3 阴道、尿道给药制剂细菌数每1g、lml 或l0cm2,不得过100CFU 。
霉菌数和酵母菌数每1g、lml 或l0cm2应小于10CFU 。
金黄色葡萄球菌、铜绿假单胞菌、白色念珠菌每1g、lml 或l0cm2,不得检出。
3 .4 直肠给药制剂细菌数每1g不得过l000CFU。
每lml 不得过100CFU 。
霉菌和酵母菌数每1g 或lml 不得过100CFU 。
金黄色葡萄球菌、铜绿假单胞菌每lg 或lml 不得检出。
3.5 其他局部给药制剂细菌数每1g、lml 或l0cm2不得过100CFU 。
霉菌和酵母菌数每1g、lml 或l0cm2不得过100CFU 。
金黄色葡萄球菌、铜绿假单胞菌每1g、lml 或l0cm2不得检出。
4.含动物组织(包括提取物)的口服给药制剂每10g 或10ml 还不得检出沙门菌。


微生物限度检查标准操作规程1 目的与适用范围本规程规定了微生物限度检查方法和操作要求。
本规程适用公司所需进行微生物限度的检查。
2 职责质量保证部负责本规程的实施。
3 内容3.1 引用标准:《中国药典》2010年版二部。
3.2 药品微生物限度检查法总则3.2.1 抽样3.2.1.1 供试品一般按批号随机抽样。
3.2.1.2 一般抽样量为检验用量(2个以上最小包装单位)的3倍量。
3.2.1.3 抽样时,凡发现有异常可疑的样品,应选有疑问的样品,但因机械损伤明显破裂的包装不得作为样品,凡已能从药品、瓶口(外盖内侧及瓶口周围)外观看出长螨、长霉、虫蛀及变质的药品,可直接判为不合格,无需要再抽样检验。
3.2.2 供试品保存供试品在检验之前,应保存在阴凉干燥处,以防供试品中的污染菌因保藏条件所引起致死、损伤或繁殖。
3.2.2.2 供试品在检验之前,应该保持原有包装状态,严禁开启,包装已开启的样品不得作为供试品。
3.2.3 检验3.2.3.1 供试品检验项目按《中国药典》2010年版二部微生物限度标准确定。
3.2.3.2 检验的全过程,均应严格遵守无菌操作,严防再污染。
3.2.3.3 除另有规定外,供试品制备成供试液后,应在均匀状态取样。
3.2.3.4 制成供试液后,应该在60分钟内注皿操作完毕。
3.2.4 培养3.2.4.1 除另有规定外,本检查法中细菌培养温度为30~35℃,霉菌、酵母菌培养温度为25~28℃,控制菌培养温度为36℃±1℃。
3.2.5 复检3.2.5.1 菌数测定不合格者应复检,控制菌检查以一次检出为准,不再复试,但应保留检出菌株一个月备查。
3.2.5.2 复试项目以不合格项目为准,作单项复试。
3.2.5.3 复试需另取同批号样品,复试2次。
3.2.5.4 复试报告,以3次测定结果的平均值报告。
3.2.6 检验报告3.2.6.1 检验报告以1g、1ml或10cm2为单位。
3.2.6.2 测定菌数报告,依3.10.2.4菌数报告规则报告。

2010版中国药典微生物限度检查法增补本适用培养基2010版2010增补本供试品制备M0008B-pH7.0氯化钠-蛋白胨缓冲液M0911B-pH6.8磷酸盐缓冲液M0912B-pH7.6磷酸盐缓冲液M0008B-pH7.0氯化钠-蛋白胨缓冲液M0001B-胰酪胨大豆肉汤M0911B-pH6.8磷酸盐缓冲液M0912B-pH7.6磷酸盐缓冲液细菌菌液制备M0005B-营养肉汤M0006B-营养琼脂0.9%无菌氯化钠溶液M0001B-胰酪胨大豆肉汤M0002B-胰酪胨大豆琼脂M0008B-pH7.0氯化钠-蛋白胨缓冲液0.9%无菌氯化钠溶液白色念珠菌菌液制备M4101B-改良马丁培养基M4102B-改良马丁琼脂培养基0.9%无菌氯化钠溶液含0.05%聚山梨酯80的0.9%无菌氯化钠溶液M4109B-沙氏萄萄糖肉汤(SDB)M4108B-沙氏葡萄糖琼脂(SDA)M0008B-pH7.0氯化钠-蛋白胨缓冲液0.9%无菌氯化钠溶液霉菌菌液制备M4102B-改良马丁琼脂培养基含0.05%聚山梨酯80的0.9%无菌氯化钠溶液M4108B-沙氏葡萄糖琼脂(SDA)M4110B-马铃薯葡萄糖琼脂(PDA)M0076B-含0.05%聚山梨酯80的pH7.0氯化钠-蛋白胨缓冲液生孢梭菌菌液制备M5601B-硫乙醇酸盐流体培养基(FT)0.9%无菌氯化钠溶液M5604B-梭菌增菌培养基M0008B-pH7.0氯化钠-蛋白胨缓冲液微生物计数法(细菌、霉菌及酵母菌计数) M0006B-营养琼脂(细菌)M4103B-玫瑰红钠琼脂(霉菌及酵母菌)M4101B-酵母浸出粉胨葡萄糖琼脂培养基(YPD)(酵母菌)M0002B-胰酪胨大豆琼脂(需氧菌)M0001B-胰酪胨大豆肉汤(需氧菌-MPN法)M4108B-沙氏葡萄糖琼脂(SDA) (霉菌和酵母菌)M4182B-沙氏葡萄糖琼脂(含抗生素) (用于霉菌和酵母菌)M4103B-玫瑰红钠琼脂(用于霉菌和酵母菌)耐胆盐革兰氏阴性菌无M2007B-肠道菌增菌肉汤M2005B-紫红胆盐葡萄糖琼脂大肠埃希氏菌M1002B-乳糖胆盐发酵培养基M1003B-曙红亚甲蓝琼脂培养基(EMB)M1004B-麦康凯琼脂培养基(MacC)M1006B-乳糖发酵培养基M2013B-麦康凯肉汤M2012B-麦康凯琼脂沙门氏菌M2002B-四硫磺酸钠亮绿培养基(TTB)基础M2004B-胆盐硫乳琼脂培养基(DHL)M2003B-沙门、志贺菌属琼脂培养基(SS)M1003B-曙红亚甲蓝琼脂培养基(EMB)M1004B-麦康凯琼脂培养基(MacC)M0009B-三糖铁琼脂培养基(TSI)M2015B-RV沙氏增菌肉汤M2010B-木糖赖氨酸脱氧胆盐琼脂(XLD)M0009B-三糖铁琼脂培养基(TSI)铜绿假单胞菌M1001B-胆盐乳糖培养基(BL)M3001B-溴化十六烷基三甲铵琼脂培养基M0006B-营养琼脂M3002B-绿脓菌素测定用培养基(PDP)基础(添加S0502)T010-氧化酶试纸M3003B-溴十六烷三甲铵琼脂基础(添加S0502)T010-氧化酶试纸金黄色葡萄球菌M0005B-营养肉汤(NB)S3401-亚碲酸钾溶液M3401B-卵黄氯化钠琼脂培养基基础(添加S3402)M3402B-甘露醇氯化钠琼脂M0006B-营养琼脂M3402B-甘露醇氯化钠琼脂M0006B-营养琼脂梭菌M5604B-梭菌增菌培养基M5602B-哥伦比亚琼脂培养基基础(添加S5601)T040-3%过氧化氢溶液M5604B-梭菌增菌培养基M5602B-哥伦比亚琼脂培养基基础(添加S5601)T040-3%过氧化氢溶液白色念珠菌M4105B-沙氏葡萄糖液体培养基M4106B-沙氏葡萄糖琼脂培养基M2509A-念珠菌显色培养基M4107A-1%吐温80-玉米琼脂培养基基础(添加S4101)M4109B-沙氏萄萄糖肉汤(SDB)M4108B-沙氏葡萄糖琼脂(SDA)M2509A-念珠菌显色培养基。

药典微生物限度标准
药典微生物限度标准是指药典(如中国药典、美国药典等)规定的药品中允许存在的微生物数量的限制。
微生物限度是为了保证药品的质量和安全所设立的,合理的微生物限度可防止和减少可能在药品生产和贮存过程中引入的微生物污染。
药典微生物限度标准通常需要考虑以下几个方面:
1. 总微生物数:药典规定了应当检测的总微生物数的限制,表示制备过程中可能存在的各种微生物的总数量。
不同类别的药品对总微生物数的限制可能有所不同。
2. 霉菌和酵母菌:霉菌和酵母菌是一类常见的微生物,它们生长迅速,可以在药品中引起变质和腐败。
因此,药典通常规定了对霉菌和酵母菌的数量限制。
3. 大肠菌群:大肠菌群是常见的肠道致病菌,它们存在于环境中,如食品、水源等,可能通过生产过程中的污染进入药品中。
药典通常规定了对大肠菌群的数量限制。
4. 特定致病菌:药典对某些具有危害性的致病菌也可能设置了数量限制,如沙门氏菌、金黄色葡萄球菌等。
药典微生物限度标准的设定是为了确保药品的质量和安全,各国的药典对微生物限度标准可能会有所不同。
生产药品时,企业需要按照药典要求的微生物限度标准进行检测,并确保药品符合规定的标准。

二、微生物限度标准(2010版药典二部附录P115,一部P88)(一)致病菌•口服制剂每g或每ml不得检出大肠埃希菌,含动物药及脏器的药品同时不得检出沙门菌•含中药原生药粉还不得检出大肠菌群(大肠杆菌)•外用每g或每ml不得检出铜绿假单胞菌(绿脓杆菌)、金黄色葡萄球菌;梭菌;大肠埃希菌(眼、鼻、呼吸)•阴道、创伤、溃疡用制剂同时不得检出破伤风杆菌。
一次为准,检出以不合格处理(二)活螨不得检出(三)细菌与霉菌1、要求无菌或标示无菌的制剂:应符合无菌检查规定3、局部给药制剂1)用于手术、烧伤或严重创伤的:应符合无菌检查规定2)用于表皮或粘膜不完整、含药材原粉的:•细菌数:≤1000 cfu /g或10cm2,≤100 cfu /ml•霉菌和酵母菌数:≤100 cfu /g、ml或10cm2•金黄色葡萄球菌、铜绿假单胞菌:不得检出/g、ml或10cm2 (各外用制剂均同)3)用于表皮或粘膜完整、含药材原粉的:细菌数:≤10 000 cfu /g或10cm2,≤100 cfu/ml霉菌和酵母菌数:≤100 cfu /g、ml或10cm2金黄色葡萄球菌、铜绿假单胞菌:不得检出/g、ml或10cm2 (各外用制剂均同)4)眼部给药制剂已全提升为无菌制剂原:细菌数:≤10 cfu /g或ml霉菌和酵母菌数:每1g或1ml不得检出金黄色葡萄球菌、铜绿假单胞菌、大肠埃希菌:每1g或1ml不得检出5)耳、鼻及呼吸道给药制剂细菌数:≤100 cfu /g、ml或10cm2霉菌和酵母菌数:≤10 cfu /g、ml或10cm2金黄色葡萄球菌、铜绿假单胞菌:不得检出/g、ml或10cm2大肠埃希菌:鼻及呼吸道给药制剂, 不得检出/ g、ml或10cm26)阴道、尿道给药制剂细菌数:≤100 cfu /g或ml霉菌和酵母菌数:≤10 cfu /g或ml金黄色葡萄球菌、铜绿假单胞菌、白色念珠菌:不得检出/g、ml或10cm27)直肠给药制剂细菌数:≤1000 cfu /g,≤100 cfu /ml霉菌和酵母菌数:≤100 cfu /g或ml金黄色葡萄球菌、铜绿假单胞菌:不得检出/g或ml8)其它局部给药制剂细菌数:≤100 cfu /g、ml或10 cm2霉菌和酵母菌数:≤100 cfu/g、ml或10cm2金黄色葡萄球菌、铜绿假单胞菌:不得检出/g、ml或10cm24、含动物组织及动物类原药材粉(蜂蜜、王浆、动物角、阿胶除外)的口服制剂:每10g或10ml还不得检出沙门氏菌5、有兼用途径制剂:应符合合途径的标准6、霉变、长螨者:以不合格论。
附录Ⅺ J 微生物限度检查法微生物限度检查法系检查非规定灭菌制剂及其原料、辅料受微生物污染程度的方法。
检查项目包括细菌数、霉菌数、酵母菌数及控制菌检查。
微生物限度检查应在环境洁净度10000级下的局部洁净度100级的单向流空气区域内进行。
检验全过程必须严格遵守无菌操作,防止再污染。
单向流空气区域、工作台面及环境应定期按《医药工业洁净室(区)悬浮粒子、浮游菌和沉降菌的测试方法》的现行国家标准进行洁净度验证。
供试品检查时,如果使用了表面活性剂、中和剂或灭活剂,应证明其有效性及对微生物无毒性。
除另有规定外,本检查法中细菌及控制菌培养温度为30℃~35℃;霉菌、酵母菌培养温度为23℃~28℃。
检验结果以1g、1ml、10g、10ml、10cm2为单位报告,特殊品种可以最小包装单位报告。
检验量检验量即一次试验所用的供试品量(g、ml或cm2)。
除另有规定外,一般供试品的检验量为10g或10ml;膜剂为100cm2;贵重药品、微量包装药品的检验量可以酌减。
要求检查沙门菌的供试品,其检验量应增加20g或20ml(其中10g用于阳性对照试验)。
检验时,应从2个以上最小包装单位中抽取供试品,膜剂还不得少于4片。
一般应随机抽取不少于检验用量(两个以上最小包装单位)的3倍量供试品。
供试液的制备根据供试品的理化特性与生物学特性,采取适宜的方法制备供试液。
供试液制备若需加温时,应均匀加热,且温度不应超过45℃。
供试液从制备至加入检验用培养基,不得超过1小时。
除另有规定外,常用的供试液制备方法如下。
1.液体供试品取供试品10ml,加pH7.0无菌氯化钠-蛋白胨缓冲液至100ml,混匀,作为1∶10的供试液。
油剂可加入适量的无菌聚山梨酯80使供试品分散均匀。
水溶性液体制剂也可用混合的供试品原液作为供试液。
2.固体、半固体或黏稠性供试品取供试品10g,加pH7.0无菌氯化钠-蛋白胨缓冲液至100ml,用匀浆仪或其他适宜的方法,混匀,作为1∶10的供试液。
药典微生物限度标准微生物限度是指在药品中允许存在的微生物数量的标准,它是评价药品质量的重要指标之一。
微生物限度标准的制定是为了保证药品的安全性、有效性和稳定性,有效地控制微生物对药品的污染和变质。
微生物限度标准的制定需要考虑药品的性质、用途、生产工艺和储存条件等因素,以确保药品在整个生产和使用过程中都能保持良好的品质。
微生物对药品的污染可能会导致药品的变质和降低药品的有效性,甚至对人体健康造成危害。
因此,制定合理的微生物限度标准对于保障药品质量和人体健康具有重要意义。
微生物限度标准通常包括细菌总数、霉菌和酵母菌的数量限度。
细菌总数是指在一定数量的制剂中存在的细菌总数的限度,它反映了药品的卫生状况和生产过程的卫生控制水平。
霉菌和酵母菌是常见的微生物污染源,它们可能会导致药品的霉变和酵败,因此对其数量也有一定的限制标准。
在药典中,针对不同类型的药品和不同的用途,制定了相应的微生物限度标准。
例如,对于口服固体制剂,其微生物限度标准要求较为严格,因为口服固体制剂在服用过程中会直接进入消化道,对人体的影响较大。
而对于外用制剂,其微生物限度标准相对较宽松,因为外用制剂在使用过程中不会直接进入人体内部,对人体的影响较小。
此外,对于注射剂和眼科制剂等高风险药品,其微生物限度标准要求更为严格,以确保药品的安全性和稳定性。
在生产过程中,严格控制微生物的污染是确保药品质量的关键步骤之一。
生产企业需要建立健全的生产管理体系,包括良好的生产环境、严格的生产操作规程、有效的清洁消毒措施等,以确保药品在生产过程中不受微生物污染。
同时,生产企业还需要建立完善的微生物检测方法和检测体系,对原辅料、中间体和成品进行全面的微生物检测,确保药品符合微生物限度标准的要求。
总之,微生物限度标准是评价药品质量的重要指标之一,对于保障药品质量和人体健康具有重要意义。
生产企业应严格遵守药典中制定的微生物限度标准,建立健全的生产管理体系,确保药品在生产和使用过程中符合微生物限度标准的要求,为人体健康提供保障。
微生物限度检查法标准操作规程一、目的:建立一个微生物限度检查标准操作规程,规范质检员的操作,保证实验的安全性、准确性。
二、范围:适用于微生物限度检查的产品。
三、责任:QC部质检员对本规程实施负责。
四、内容1.检验依据:《中国药典》2010年版二部。
2. 简述2.1.微生物限度检查法系检查非规定灭菌制剂及其原料、辅料受微生物污染程度的方法。
检查项目包括细菌数、霉菌数、酵母菌数及控制菌数检查。
2.2.微生物限度检查应在环境洁净度10000级下的局部洁净度100级的单向流空气区域内进行。
检验全过程必须严格遵守无菌操作,防止再污染,防止污染的措施不得影响供试品中微生物的检出。
单向流空气区域、工作台面及环境应定期按《医药工业洁净室(区)悬浮粒子、浮游菌和沉降菌的测试方法》的现行国家标准进行洁净度验证。
2.3.供试品检查时,如果使用了表面活性剂、中和剂或灭活剂,应证明其有效性及对微生物无毒性。
2.4.除另有规定外,本检查法中细菌及控制菌培养温度为30~35℃;霉菌、酵母菌培养温度为23~28℃2.5.检验结果以1g、1ml、10g、10ml或10cm2为单位报告,特殊品种可以最小包装单位报告。
3.设备、仪器3.1.设备:3.1.1.洁净实验室:微生物限度检查应有单独的洁净实验室,每个洁净实验室应有独立的净化空气系统。
结构和要求:洁净室应采光良好,避免潮湿、远离厕所及污染区。
操作间与缓冲间应有样品传递窗,出入操作间和缓冲间的门不应直对。
洁净实验室内应六面光滑平整,能耐受清洗消毒。
墙壁与地面、天花板连接处无缝隙,不留死角。
操作间不应安装下水道。
洁净实验室内的照明灯应嵌装在天花板内,室内光照应分布均匀,光照度不低于300LX。
●温度、湿度:洁净实验室内温度应控制在18~26℃,相对湿度最好在40%~60%。
●操作间:操作间应安装空气除菌过滤层流装置。
洁净度不应低于10000级,局部洁净度为100级(或放置同等级净化工作台)。
《中国药典》2010年版二部附录XI J (附录115页)《微生物限度检查法》微生物限度标准非无菌药品的微生物限度标准是基于药品的给药途径及对患者健康潜在的危害而制订的。
药品的生产、贮存、销售过程中的检验,原料及辅料的检验,新药标准制订,进口药品标准复核,考察药品质量及仲裁等,除另有规定外,其微生物限度均以本标准为依据。
1.制剂通则、品种项下要求无菌的制剂及标示无菌的制剂应符合无菌检查法规定。
2.口服给药制剂细菌数每1g不得过l000CFU 。
每lml 不得过100CFU 。
霉菌和酵母菌数每lg或lml 不得过100CFU 。
大肠埃希菌每1g 或lml不得检出.3 .局部给药制剂3.1用于手术、烧伤及严重创伤的局部给药制剂应符合无菌检查法规定。
3.2 耳、鼻及呼吸道吸入给药制剂细菌数每1g、lml 或l0cm2,不得过100CPU 。
霉菌和酵母菌数每1g、lml 或l0cm2,不得过10CPU 。
金黄色葡萄球菌、铜绿假单胞菌每1g、lml 或l0cm2不得检出。
大肠埃希菌鼻及呼吸道给药的制剂,每1g、lml 或l0cm2,不得检出。
3.3 阴道、尿道给药制剂细菌数每1g、lml 或l0cm2,不得过100CFU 。
霉菌数和酵母菌数每1g、lml 或l0cm2应小于10CFU 。
金黄色葡萄球菌、铜绿假单胞菌、白色念珠菌每1g、lml 或l0cm2,不得检出。
3 .4 直肠给药制剂细菌数每1g不得过l000CFU。
每lml 不得过100CFU 。
霉菌和酵母菌数每1g 或lml 不得过100CFU 。
金黄色葡萄球菌、铜绿假单胞菌每lg 或lml 不得检出。
3.5 其他局部给药制剂细菌数每1g、lml 或l0cm2不得过100CFU 。
霉菌和酵母菌数每1g、lml 或l0cm2不得过100CFU 。
金黄色葡萄球菌、铜绿假单胞菌每1g、lml 或l0cm2不得检出。
4.含动物组织(包括提取物)的口服给药制剂每10g 或10ml 还不得检出沙门菌。
2010年版《中国药典》二部制剂通则口服溶液剂:药物溶解于适宜溶剂中制成供口服的澄清液体制剂。
滴剂:用适宜的量具以小体积或以滴计量的口服溶液剂、口服混悬剂、口服乳剂的液体制剂。
生产与贮藏期间应符合下列规定:1.口服溶液剂的溶剂常用纯化水。
2.根据需要可加入适宜的附加剂,如防腐剂、分散剂、助悬剂、增稠剂、助溶剂、润湿剂、缓冲剂、稳定剂、乳化剂、矫味剂以与色素等。
其品种与用量应符合国家标准的有关规定,不影响产品的稳定性,并避免对检验产生干扰。
3.不得有发霉、酸败、变色、异物、产生气体或其他变质现象。
4.除另有规定外,应密封、遮光贮存。
除另有规定外,口服溶液剂应进行一下相应检查。
装量:除另有规定外,单剂量包装的口服溶液剂装量,应符合下列规定。
取供试品10支,分别将内容物倾尽,测定其装量,每支装量均不得少于其标示量。
微生物限度:照微生物限度检查法(2010年版《中国药典》附录XI J)检查,应符合规定。
附录XI J微生物限度检查法检验量:一次试验所用的供试品量。
除另有规定外,一般供试品的检验量为10g或10ml。
要求检查沙门菌的供试品,其检验量应增加20g或20ml。
检验时,应从2个以上最小包装单位中抽取供试品。
一般应随机抽取不少于检验用量(两个以上最小包装单位)的3倍量供试品。
微生物限度标准:口服给药制剂细菌数:每1ml不得过100cfu。
霉菌和酵母菌数:每1ml不得过100cfu。
大肠埃希菌:每1ml不得检出。
防腐剂用量限度:2010版《中国药典》附录I K糖浆剂。
山梨酸和苯甲酸的用量不得超过0.3%,(其钾盐、钠盐的用量分别按酸计算)。
羟苯酯类的用量不得超过0.05%。
2010年版《中国药典》附录XIX N 抑菌剂效力检查法指导原则抑菌剂效力检查法是用于测定灭菌、非灭菌制剂中抑菌剂的活性,以评价最终产品的抑菌效力,同时也可用于指导生产企业在研发阶段制剂中抑菌剂浓度的确定。
产品分类属于3类:口服非固体制剂(非抗酸剂)。
《中国药典》2010年版二部附录XI J (附录115页)
《微生物限度检查法》
微生物限度标准
非无菌药品的微生物限度标准是基于药品的给药途径及对患者健康潜在的危害而制订的。
药品的生产、贮存、销售过程中的检验,原料及辅料的检验,新药标准制订,进口药品标准复核,考察药品质量及仲裁等,除另有规定外,其微生物限度均以本标准为依据。
1.制剂通则、品种项下要求无菌的制剂及标示无菌的制剂应符合无菌检查法规定。
2.口服给药制剂
细菌数每1g不得过l000CFU 。
每lml 不得过100CFU 。
霉菌和酵母菌数每lg或lml 不得过100CFU 。
大肠埃希菌每1g 或lml不得检出.
3 .局部给药制剂
3.1用于手术、烧伤及严重创伤的局部给药制剂应符合无菌检查法规定。
3.2 耳、鼻及呼吸道吸入给药制剂
细菌数每1g、lml 或l0cm2,不得过100CPU 。
霉菌和酵母菌数每1g、lml 或l0cm2,不得过10CPU 。
金黄色葡萄球菌、铜绿假单胞菌每1g、lml 或l0cm2不得检出。
大肠埃希菌鼻及呼吸道给药的制剂,每1g、lml 或l0cm2,不得检出。
3.3 阴道、尿道给药制剂
细菌数每1g、lml 或l0cm2,不得过100CFU 。
霉菌数和酵母菌数每1g、lml 或l0cm2应小于10CFU 。
金黄色葡萄球菌、铜绿假单胞菌、白色念珠菌每1g、lml 或l0cm2,不得检出。
3 .
4 直肠给药制剂
细菌数每1g不得过l000CFU。
每lml 不得过100CFU 。
霉菌和酵母菌数每1g 或lml 不得过100CFU 。
金黄色葡萄球菌、铜绿假单胞菌每lg 或lml 不得检出。
3.5 其他局部给药制剂
细菌数每1g、lml 或l0cm2不得过100CFU 。
霉菌和酵母菌数每1g、lml 或l0cm2不得过100CFU 。
金黄色葡萄球菌、铜绿假单胞菌每1g、lml 或l0cm2不得检出。
4.含动物组织(包括提取物)的口服给药制剂每10g 或10ml 还不得检出沙门菌。
5.有兼用途径的制剂应符合各给药途径的标准。
6.霉变、长螨者以不合格论。
7.原料及辅料参照相应制剂的微生物限度标准执行。
注本检查法中白色念珠菌检查所描述该菌在念珠菌显色培养基上的菌落特征,是指该菌在法国科玛嘉公司提供的科玛嘉念珠菌显色培养基上生长的菌落特征。
给出这一信息是为了方便本方法的使用者,并不表示对该公司产品的认可.若其他等效产品具有相同的效果,那么也可使用其他等效的产品。