原子团簇的稳定结构和幻数-引言
- 格式:ppt
- 大小:1.03 MB
- 文档页数:20


原子团簇的稳定结构和幻数是指团簇中原子数目的影响。
当团簇中含有某些特定原子数目时,团簇会特别稳定,这些特定的原子数目被称为幻数。
例如,当团簇中原子数目为2、8、20、40、58、114时,团簇会达到相对稳定的状态。
原子团簇介于原子和宏观凝聚物质之间,其结构和性质随所含原子数目而变化。
由于电子壳层的存在,使得某些特定原子数目的团簇具有相对稳定结构。
例如,当团簇中原子数目为2、8、20、40、58、114时,电子壳层结构能够使团簇中的原子更好地结合在一起,形成相对稳定的结构。


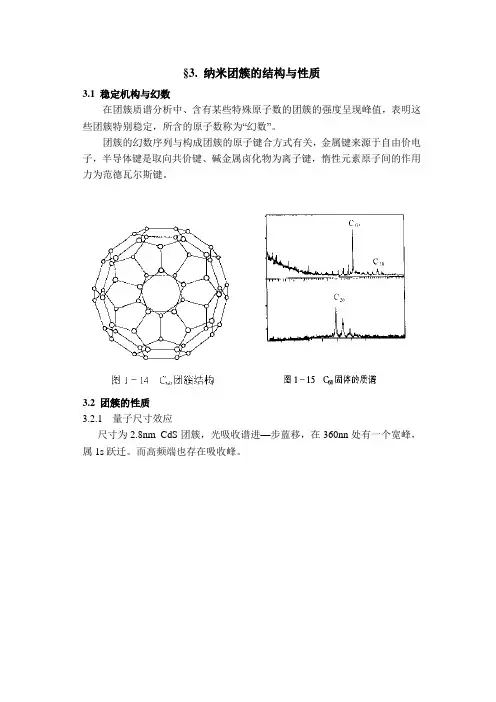
§3. 纳米团簇的结构与性质
3.1 稳定机构与幻数
在团簇质谱分析中、含有某些特殊原子数的团簇的强度呈现峰值,表明这些团簇特别稳定,所含的原子数称为“幻数”。
团簇的幻数序列与构成团簇的原子键合方式有关,金属键来源于自由价电子,半导体键是取向共价键、碱金属卤化物为离子键,惰性元素原子间的作用力为范德瓦尔斯键。
3.2 团簇的性质
3.2.1 量子尺寸效应
尺寸为2.8nm CdS团簇,光吸收谱进—步蓝移,在360nn处有一个宽峰,属1s跃迁。
而高频端也存在吸收峰。
实验表明纳米尺寸的半导体团簇具有可贵的光学性质,即分立的能级跃迂,并与团簇尺寸和形状密切有关。
3.2.2 电子性质
(1)下图给出了钾团簇电离势随n的变化”,可以看出直至n接近100,电离势具有与团族幻数相对应的峰值,在某一壳层连续填充的过程中,电离势近似一常数,但在每一个壳层填满时,电离势发生突变。
(2)带负电铜簇Cu-(n=1-410)进行紫外光电子谱实验,通过观察光电子发射可以直接估计出相应中性团族的电子亲和势。
下图是有各种原子数的铜团簇Cu n-的光电子谱。
3.2.3光学性质
金属团簇对光的响应具有和单个原于及大块固体均不相向的特征。
下图示出尺寸分别为2nm,14nm和20nm铜闭簇嵌埋于氟化理基体中的光
吸收谱,下表给出了实验结果。
随着团簇尺寸增加.峰位红移.峰展宽。

§3. 纳米团簇的结构与性质
3.1 稳定机构与幻数
在团簇质谱分析中、含有某些特殊原子数的团簇的强度呈现峰值,表明这些团簇特别稳定,所含的原子数称为“幻数”。
团簇的幻数序列与构成团簇的原子键合方式有关,金属键来源于自由价电子,半导体键是取向共价键、碱金属卤化物为离子键,惰性元素原子间的作用力为范德瓦尔斯键。
3.2 团簇的性质
3.2.1 量子尺寸效应
尺寸为2.8nm CdS团簇,光吸收谱进—步蓝移,在360nn处有一个宽峰,属1s跃迁。
而高频端也存在吸收峰。
实验表明纳米尺寸的半导体团簇具有可贵的光学性质,即分立的能级跃迂,并与团簇尺寸和形状密切有关。
3.2.2 电子性质
(1)下图给出了钾团簇电离势随n的变化”,可以看出直至n接近100,电离势具有与团族幻数相对应的峰值,在某一壳层连续填充的过程中,电离势近似一常数,但在每一个壳层填满时,电离势发生突变。
(2)带负电铜簇Cu-(n=1-410)进行紫外光电子谱实验,通过观察光电子发射可以直接估计出相应中性团族的电子亲和势。
下图是有各种原子数的铜团簇Cu n-的光电子谱。
3.2.3光学性质
金属团簇对光的响应具有和单个原于及大块固体均不相向的特征。
下图示出尺寸分别为2nm,14nm和20nm铜闭簇嵌埋于氟化理基体中的光
吸收谱,下表给出了实验结果。
随着团簇尺寸增加.峰位红移.峰展宽。


平面C n B3(n=1~8)团簇的结构与稳定性的探索1 引言在轻质半导体中,碳化硼是硬度仅次于金刚石和立方氮化硼的一种材料。
碳化硼具有对中子的高俘获性,高强度,较高的杨氏模量和较高的热稳定性。
由于硼碳材料(纳米线,纳米弹簧和纳米带等)具有耐高温,耐酸,高硬度和高强度等特性,这类材料在核聚变反应器和航空器等领域有潜在的应用,这说明了硼碳材料是有应用前景的材料。
因此,近几十年来硼碳材料被广泛地研究。
近年来,硼碳团簇已经成为许多实验和理论研究的热门话题。
实验检测只能间接地提供较小的混合硼碳团簇结构信息。
尽管量子化学计算和分子动力学模拟能够提供团簇的直接结构信息,但是团簇稳定的几何结构的数目是随团簇中的原子个数的增加而呈指数增加。
所以,全局最低能量结构的确定是一个具有挑战性的任务。
前人的理论计算预测了具有平面四配位碳,平面五配位碳和平面六配位碳等有趣的硼碳团簇的结构。
到目前为止,有关硼碳混合团簇的结构的实验结果报道很少。
因而,对硼碳混合二元团簇的计算研究是重要而有趣的,也很有挑战性。
2 计算方法首先,采用B3LYP/6-31G(d)理论方法对二重态和四重态的平面C n B3 (n=1-8)团簇的线性结构、支链结构、环状结构及带有支链的环状结构等四种没有对称限制的结构样式进行了几何结构优化。
然后,在B3LYP/6-311+G(d)理论水平下对CB3的12个能量较低的异构体中和每一个C n B3 (n=2-8)分子的30个异构体进行进一步的几何优化。
为了检测这些异构体的稳定性,进行了振动频率分析。
为了获得更精确地能量次序,在CCSD(T)/6-311+G(d)理论水平上对CB3的12个能量较低的异构体中和每一个C n B3 (n=2-8)分子的30个异构体进行了能量校正。
所有的计算都是在Gaussian 09软件包上完成[39]。
3 结果与讨论经过优化的C n B3 (n=2~8)每个团簇的10个异构体的几何结构显示在图1中。

Ⅳ族元素团簇结构及稳定性的理论研究的开题报告一、研究的背景和意义地球上的元素由化学元素周期表所描述的118种元素构成,其中Ⅳ族元素包括碳、硅、锗、锡和铅等五种元素。
这些元素具有重要的地球化学、材料科学、生命科学和环境科学等方面的研究价值。
因此,对Ⅳ族元素的物理化学性质、结构以及其在各种体系中的作用机理进行深入研究和探究,具有十分重要的理论研究意义和实际应用价值。
目前,科学家们已经将Ⅳ族元素的超晶格结构、纳米晶等微观结构进行了广泛的研究,这些研究为理解Ⅳ族元素的物理化学特性以及其在材料科学、生命科学、环境科学等领域的应用奠定了基础。
然而,对于Ⅳ族元素的团簇结构以及其稳定性的研究相对较少,而这些微小的团簇结构在实际应用中很可能发挥着重要的角色,因此对其进行深入研究与探究,对于理解Ⅳ族元素的整体性质和在各种体系中的作用机理,具有重要的理论意义和实际应用价值。
二、研究的目的和内容本研究的主要目的是:利用量子化学计算方法对Ⅳ族元素的团簇结构及其稳定性进行透彻的理论研究,并且通过大量计算得到具有实际意义的结论,期望能为其在材料科学、生命科学、环境科学等方面的应用提供更深入的理论基础和研究方向。
本研究将首先从理论上探讨Ⅳ族元素的团簇结构的组成规律以及其形成机制,然后对于不同形式的团簇结构进行计算研究,探究其稳定性、能量结构、几何结构和电子结构等方面的性质,并且结合实验数据进行验证和分析,从而得到具有实际意义的研究成果。
三、研究的方法和步骤本研究将采用量子化学计算方法,特别是密度泛函理论(DFT)方法,对Ⅳ族元素的团簇结构进行计算研究。
具体方法步骤如下:1. 确定研究对象——Ⅳ族元素的团簇结构;2. 选择合适的计算方法和软件,建立符合实际物理条件的计算模型;3. 进行模型优化计算,捕捉可能的全局最小能量构型;4. 对计算结果进行几何构型的优化和频率分析,确定结构是否为局部或全局能量最低点;5. 在确定了结构和能量之后,进一步进行电子结构分析,研究其电子亲和性、电子结构、反应活性等方面的性质;6. 结合实验数据对计算结果进行验证和分析。

团簇及掺杂团簇的研究现状及意义原子团簇和分子团簇,简称为团簇(Cluster);团簇这一名词是Cotton在1996年提出的,并认为团簇是具有金属-金属键的多核化合物。
团簇由几个乃至上千个原子、分子或离子通过物理或化学结合力组成的相对稳定的微观和亚微观聚集体,团簇的空间尺度大约在几埃至几百埃,其物理以及化学性质随所含原子数的变化而变化。
团簇的许多性质不同于单个的原子或分子,也不同于固体或液体,并且也不能从单体和体相材料的性质作简单的线性外延和内插来得到。
因此,团簇被看作是介于原子分子以及宏观固体之间物质结构的新层次,称之为物质的“第五态”,它是各种物质由原子分子向体相物质转变的过渡态,也可说它是代表了凝聚态物质的初始状态,团簇的研究有利于我们认识由单个原子向大块凝聚物过渡时性质的变化规律。
团簇广泛存在于自然界与人类的实践活动中,涉及的许多现象如燃烧、晶体生长、催化、成核和凝固、相变与临界现象、薄膜形成、溶胶和溅射等可构成物理和化学的一个交汇点。
况且,在团簇中还出现了些新的物理现象,例如壳层结构与幻数、液相与固相并存与转化、表面等离子激发、磁性增强、同位素效应以及金属非金属转变等等。
因而对团簇的研究将带动凝聚态物理、表面物理和化学、原子分子物理和化学动力学的发展。
团簇作为介于气态与固态之间的一种过渡态,对其形成和运动规律的研究,不仅为发展和完善原子间结合理论以及各种固体和大分子形成规律提供了合适的对象,也是在实验条件下对大气烟雾和溶胶、宇宙分子和尘埃、云层的形成和发展等的一种模拟,可为气侯人工调节、大气污染控制和天体演化的研究提供线索,丰富了生命科学、大气科学和宇宙科学学科的内容。
另外,团簇的理论研究也促进了理论物理和计算物理的发展。
团簇在空间上是有限尺度的,零维至三维的模型系统可通过对其几何结构的选择来提供。
在团簇的理论研究中,所开发出的一些计算方法也可进一步的推广到有机分子、生物大分子以及固体材料等复杂的系统的计算模拟中。
原子核幻数结构新解析林文业关键词:原子核;幻数;结构。
本人经过十多年的探索研究,找到了物质结构及相互作用的统一规律,并写成了两本书出版(郑州大学出版社),书名叫《现代数学与量子力学》(2010年)和《相互作用统一理论》(2014年),下面我把书中物理学大统一理论的内容拿出来供大家分享。
原子核幻数结构的最新解析上面我们对原子核外的电子分布已有一个清析的认识,那么原子核的结构规律又是怎样的?由前面的论述可知,物质中心核的形成是物质空间的若干个量子态由第2态经基态裂变到第n (2n >)态形成新物质空间,再填充微粒子形成的。
(1) 核子结构的性质核子一般可能存在以下几种结构形式① 中子()()()()216164035432⨯⋅-⋅-⋅=⋅⋅⋅n n n n , 质子()()()216321035432⨯⋅-⋅⋅=⋅⋅⋅n n n n ,1表示填充的粒子为光类子,其余则表示填充的粒子为1/2μ类中微子。
① 质子或反质子()()()[]10985322222221⋅⋅⋅⋅⋅⋅,中子()()()[]10985322222222⋅⋅⋅⋅⋅⋅ 填充的粒子为正负电子对。
③ 核子()()3543216000⋅⋅⋅=⋅⋅⋅n n n n ,填充的粒子为正反μ类中微子对。
④ 核子()()354322000⋅⋅⋅=⋅⋅⋅n n n n ,填充的粒子为正反π粒子对。
根据“间杂填充不湮灭准则”,在上述4种结构形式中还应间杂填充部分其它微粒子,粒子才稳定。
从上面这些结构形式,不难看出核子结构的性质: 核子若除了电性,其最外层量子态必然至少填充两个性质互为相反的正反微粒子。
如果定义填充量子态最外侧为正微粒子的核子为左核子,那么定义填充量子态最外侧为反微粒子的核子则为右核子。
可见核子可分为左核子和右核子两种。
根据泡利不相容原理, 原子核的核子填充有这样的组对: 质子—中子对和左中子—右中子对(即简称中子—中子对)。
(2) 核子组成原子核的规则:原子核的形成是原子核空间的s 个量子态由第2态经基态裂变成新物质空间的32s 个轨道基态,和32s 个核心自旋态,以及由填充壳层产生的()()112---m m 至()()112-+-m m 个壳层自旋态(其中m 为填充层数,1m >,第一个填充层只有1个壳层自旋态)。
CnAl(n=2-11)团簇的结构特征与稳定性
王艳宾;马文瑾;张静;武海顺
【期刊名称】《物理化学学报》
【年(卷),期】2007(23)6
【摘要】采用密度泛函理论(DFT)的B3LYP方法,在6-311++G**水平上对CnAl(n=2-11)团簇的几何构型和电子结构进行了结构优化和振动频率计算.结果表明,n=2的CnAl团簇基态结构为Al原子与两个C原子相连形成的环状结构,n=3-11均为Al原子端基配位的线状结构.通过对基态结构的能量分析,得到了n为偶数的CnAl团簇比n为奇数团族稳定的结论.
【总页数】4页(P873-876)
【作者】王艳宾;马文瑾;张静;武海顺
【作者单位】山西师范大学化学与材料科学学院,山西,临汾,041004;山西师范大学化学与材料科学学院,山西,临汾,041004;山西师范大学化学与材料科学学院,山西,临汾,041004;山西师范大学化学与材料科学学院,山西,临汾,041004
【正文语种】中文
【中图分类】O641
【相关文献】
Al2带电团簇的结构与稳定性的量子化学研究 [J], 宋翔
Al+小团簇结构的几何特征与稳定性 [J], 马文瑾;宋翔;张献明;武海顺
Al2(n=1-10)团簇的结构特征与稳定性 [J], 马文瑾;张献明;许小红;王艳宾;武
海顺
4.CoAlN(N=2-11)团簇基态结构的稳定性和磁性 [J], 王献伟;姚建刚;赵高峰
5.第一性原理研究Au_n(n=2-11)团簇的结构和稳定性 [J], 张文庆;任晓燕;刘亚明;赵高峰
因版权原因,仅展示原文概要,查看原文内容请购买。