分子模拟实验报告分子光谱模拟
- 格式:docx
- 大小:215.54 KB
- 文档页数:6


分子动力学模拟可以用来模拟红外光谱,这主要涉及到对分子振动模式的模拟。
以下是一些具体的方法和步骤:
选择合适的模型:在模拟过程中,首先需要选择一个合适的模型来描述分子间的相互作用。
这通常涉及到对分子间力场的确定,以及确定分子间的相互作用参数。
设置模拟参数:在模拟过程中,需要设置适当的参数,包括温度、压力、时间步长等。
这些参数的选择将影响到模拟结果的准确性和可靠性。
进行模拟:在设置好参数后,就可以开始进行模拟了。
分子动力学模拟可以通过各种软件包来进行,如LAMMPS、NAMD等。
这些软件包可以通过模拟分子的运动轨迹来模拟分子的振动模式。
分析模拟结果:在模拟结束后,需要对结果进行分析。
可以通过计算分子的振动频率、振动模式等来分析分子的红外光谱。
这些结果可以与实验结果进行比较,以验证模型的准确性和可靠性。
总的来说,分子动力学模拟可以用来模拟红外光谱,这有助于深入了解分子的结构和性质,从而为实验研究提供理论支持。
但需要注意的是,由于分子动力学模拟的计算量较大,因此需要进行合理的参数选择和优化,以提高模拟的效率和准确性。

分子模拟中的拉曼光谱模拟引言分子模拟是通过建立分子的精细计算机模型来研究分子结构、动态、能量等性质的一种方法。
而拉曼光谱是研究分子振动状态的非常重要的实验手段。
因此,利用计算机模拟来模拟拉曼光谱的技术逐渐成为了分子模拟领域的一个重要研究课题。
分子振动和拉曼光谱分子振动是分子内原子相对于核心位置所做的振动。
这些振动分为三种类型:拉伸、屈曲和扭曲振动。
这些振动的性质可以通过拉曼光谱来研究。
拉曼光谱是通过照射样品后收集散射光来得到的,这种散射光与入射光的频率相差数千兆赫。
这种频率差异称为拉曼位移,并可以作为识别分子结构和确定化学环境的重要参数。
例如,两个相同化学成分但不同构化的分子会有不同的拉曼光谱,因为它们的振动能量是不同的。
拉曼光谱模拟方法计算机模拟最初被用于研究分子物理和化学性质。
随着计算机技术的发展和算法的提高,人们开始尝试将计算机模拟和拉曼光谱相结合,从而模拟分子的振动状态和拉曼光谱。
目前,有两种主要的方法用于模拟拉曼光谱:量子力学计算和分子动力学计算。
量子力学计算采用基于量子力学的方法来计算分子的振动状态和拉曼光谱。
分子动力学方法则是通过模拟分子的运动来计算振动和拉曼光谱。
分子动力学方法的优势在于能够考虑分子及其周围环境的宏观行为,从而更准确地描述分子系统的振动状态和拉曼光谱。
分子动力学方法一般基于牛顿动力学方程,使用分子的力场和初始条件来模拟分子的振动。
在计算过程中,分子的位置和速度被迭代地计算,直到达到平衡态,振动和拉曼光谱得到计算和分析。
在分子动力学方法中使用的力场通常包括分子的势能函数和力常数。
势能函数描述了分子内部的化学键、角、二面角等本质特征以及它们之间的作用。
力常数描述了物理作用力,例如库仑力、范德华力和键的刚度等。
最后,通过将模拟结果与实验结果进行比较,可以调整模型参数并优化模型,从而更准确地模拟分子振动状态和拉曼光谱。
结论分子模拟中的拉曼光谱模拟是一种重要的研究课题。
通过模拟分子的振动状态和拉曼光谱,可以更准确地研究分子结构和化学环境的性质。

分子光谱实验报告1. 引言分子光谱是研究分子结构和分子振动、转动等动力学过程的重要实验手段之一。
通过测量物质在各个波长下的吸收、散射或发射光谱,可以得到关于分子能级、分子结构、分子间相互作用等信息。
本实验旨在通过分子光谱实验,学习光谱测量原理和方法,并利用可见光和红外光谱仪测量样品的吸收光谱和发射光谱。
2. 实验原理2.1 分子光谱的基本原理根据量子力学的基本原理,分子能量是量子化的,通过吸收或发射辐射能量的方式来完成能级之间的转变。
分子在外界激发下,能级之间会发生跃迁,从而吸收或发射光谱。
根据吸收、散射或发射光谱的特点,可以了解分子的结构、振动和转动等特性。
2.2 分子光谱实验仪器本实验主要使用可见光和红外光谱仪进行测量。
可见光谱仪通过对可见光的分光,得到物质在不同波长下的吸收光谱。
红外光谱仪则通过对红外光的分光,得到物质在红外光波段下的吸收光谱。
2.3 分子光谱实验步骤1.样品的制备:将待测样品溶解于适当的溶剂中,制备成测试溶液。
2.可见光谱仪实验步骤:–将一个未知浓度的样品溶液倒入样品池。
–打开可见光谱仪,调整工作波长范围和光强度。
–测量样品吸收光谱。
3.红外光谱仪实验步骤:–将样品涂在红外光谱仪的窗口上。
–打开红外光谱仪,调整工作波长范围和分辨率。
–测量样品吸收光谱。
3. 实验结果与分析3.1 可见光谱实验结果在可见光谱仪的测量中,我们选取了几个不同溶液样品,测量了它们在可见光波段下的吸收光谱。
根据测量结果,我们可以观察到样品在不同波长下的吸收情况,并根据波长对应的颜色,初步判断样品中可能存在的色素成分。
3.2 红外光谱实验结果在红外光谱仪的测量中,我们涂抹了样品在红外光谱仪的窗口上,测量了它们在红外波段下的吸收光谱。
根据测量结果,我们可以观察到样品在不同波数下的吸收情况,并通过对比基础物质的红外吸收峰位置,初步判断样品中可能存在的官能团。
4. 结论通过本次分子光谱实验,我们学习了分子光谱的基本原理和方法,并且使用可见光和红外光谱仪对样品进行了测量。

分子模拟实验溶剂化效应和红外光谱的模拟实验内容介绍:本次实验主要内容是溶剂化效应和红外光谱模拟。
何谓溶剂化效应,通常量子化学的研究都是在真空中、绝对零度下、气相分子的性质,实际上大多数化学物质和过程都存在于介质(如各种溶剂)中,与孤立的气相分子相比,溶剂对溶质分子的性质及其参与的化学反应,都有可能有非常重要的影响。
不同的溶剂不仅可以影响溶质分子的结构、反应平衡、反应速率,甚至可以改变反应进程和机理,得到不同的产物或产率。
将这些影响称之为“溶剂化效应”。
对溶剂化效应的模拟有三种模型:显式溶剂化模型、隐式溶剂化模型、显式隐式结合模型。
显式模型主要是在溶质分子中加入真正的溶剂分子再进行优化模拟;隐式模型是连续介质模型,溶剂对溶质分子的作用称为反应场,通过场概念运用迭代方法的计算,直至自洽,称自洽反应场方法(SCRF),其中又包括:Onsager模型、极化连续介质模型(PCM)。
实验中,我们以反应F- +CH3F = FCH3 +F-为例,进行溶剂化模型的建立以及反应优化计算。
红外光谱模拟是分子光谱模拟的重要一项。
分子的红外光谱是分子振动的反映,振动频率对应于红外光谱的一个谱峰,振子强度(由于振动而引起的分子偶极矩的变化)相应于光谱峰的高度。
谱峰的高度则是由于诸如热效应等引起的展宽,与分子本身的振动性质关系不大,因此模拟分子的红外光谱,首先需要对分子进行振动频率分析。
计算红外光谱时有以下几个原则:1、必须采用优化的分子结构;2、结构优化和频率计算必须采用同一理论水平;3、理论计算的频率为谐振动频率,一般偏高;4、理论计算的振子强度和实验峰高不具有可比性。
实验中,我们以H2O分子为对象,作红外光谱的模拟计算。
实验要求:1、掌握溶剂化效应的概念和溶剂化模型,能做溶剂化效应对反应进程的模拟;1、理解红外光谱的概念和模拟,作出不同优化方法下的H2O分子的红外光谱图,并比较分析。
实验一:溶剂化效应(1)、反应F- +CH3F = FCH3 +F- 是一个典型的有机反应S N2反应,反应在气相进行时,首先形成一个“中间体”,这是一个简单的符合过程,不存在过渡态。


分子模拟实验报告分子光谱模拟分子光谱模拟实验报告摘要:本实验采用分子模拟的方法,通过计算机模拟的手段,研究了分子光谱。
通过构建分子模型、选择适当的计算方法和参数,得到了分子的能级结构和光谱。
实验结果表明,分子模拟可以较好地模拟分子的能级和光谱。
这种方法可以为分子光谱的研究提供一种新的途径。
1.引言分子光谱是研究分子内部能级和分子结构的重要手段。
传统的实验方法繁琐且成本较高,分子模拟则是一种新的研究手段,可以通过计算机模拟的方法得到分子的能级结构和光谱。
本实验旨在通过分子模拟的方法,研究分子的光谱现象,并探讨模拟方法的准确性和适用性。
2.实验方法2.1分子模型的构建2.2计算方法和参数的选择选择适当的计算方法和参数对于分子模拟的准确性和有效性具有重要意义。
本次实验采用量子力学方法进行计算,选择了Hartree-Fock方法作为计算方法,并设置了合适的收敛阈值和基组。
2.3能级结构的计算通过计算机程序,对构建的分子模型进行能级结构的计算。
通过求解Schrödinger方程,可以得到分子的不同能级及其能量。
2.4光谱的模拟在能级结构的基础上,模拟分子的光谱现象。
根据波长、频率和吸收强度的关系,得到分子的吸收光谱图和发射光谱图。
3.实验结果与分析3.1能级结构的计算结果通过计算机程序,得到了水分子的能级结构。
结果显示,水分子的基态电子能级为X^1A1,第一激发态能级为A^1B1、各能级的能量差异较小,符合分子光谱的特点。
3.2光谱的模拟结果根据能级结构,模拟了水分子的吸收光谱和发射光谱。
吸收光谱图显示,在不同波长范围内,水分子的吸收强度存在明显的吸收峰,这与实验观测结果一致。
发射光谱图显示,水分子在受激条件下会发出特定波长的光,这也符合实验观测结果。
4.结论通过分子模拟实验,我们成功地模拟了水分子的能级结构和光谱现象。
实验结果表明,分子模拟可以较好地模拟分子的能级和光谱,为分子光谱的研究提供了一种新的途径。
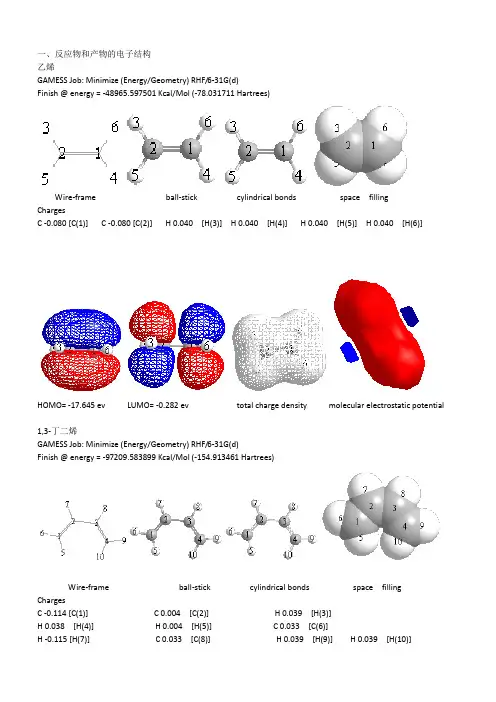
一、反应物和产物的电子结构乙烯GAMESS Job: Minimize (Energy/Geometry) RHF/6-31G(d)Finish @ energy = -48965.597501 Kcal/Mol (-78.031711 Hartrees)Wire-frame ball-stick cylindrical bonds space fillingChargesC -0.080 [C(1)] C -0.080 [C(2)] H 0.040 [H(3)] H 0.040 [H(4)] H 0.040 [H(5)] H 0.040 [H(6)]HOMO= -17.645 ev LUMO= -0.282 ev total charge density molecular electrostatic potential1,3-丁二烯GAMESS Job: Minimize (Energy/Geometry) RHF/6-31G(d)Finish @ energy = -97209.583899 Kcal/Mol (-154.913461 Hartrees)Wire-frame ball-stick cylindrical bonds space fillingChargesC -0.114 [C(1)] C 0.004 [C(2)] H 0.039 [H(3)]H 0.038 [H(4)] H 0.004 [H(5)] C 0.033 [C(6)]H -0.115 [H(7)] C 0.033 [C(8)] H 0.039 [H(9)] H 0.039 [H(10)]HOMO= -15.068 ev LUMO= -3.719ev total charge density molecular electrostatic potential环己烯GAMESS Job: Minimize (Energy/Geometry) RHF/6-31G(d)Finish @ energy = -146221.8807 Kcal/Mol (-233.019592 Hartrees)Wire-frame ball-stick cylindrical bonds space fillingChargesC -0.059 [C(1)] C -0.059 [C(2)] H -0.073 [H(3)] C -0.073 [C(4)]C -0.082 [C(5)] H 0.030 [H(6)] H -0.082 [H(7)] C 0.030 [C(8)]H 0.052 [H(9)] H 0.052 [H(10)] H 0.052 [H(11)] H 0.052 [H(12)]H 0.040 [H(13)] C 0.040 [C(14)] H 0.040 [H(15)] H 0.040 [H(16)]HOMO= -14.895 ev LUMO= 1.618 ev total charge density molecular electrostatic potential从分子轨道的对称性来看,乙烯的HOMO轨道关于镜面呈对称,LUMO反对称,丁二烯的HOMO反对称,LUMO 对称,环己烯HOMO对称,LUMO反对称。

分子模型实验报告篇一:分子模拟实验实验报告生物大分子分子模拟实验作业——生物大分子一、实验部分12-3-1获得PDB号为“1HCK”的蛋白(human-cyclin-dependent kinase 2,i,e.,CKD2和ATP的结合晶体结构),并采用不同的模型观察其特点①分别用卡通模型和丝带模型显示生物大分子结构,并用球棍模型、棒状模型显示其中小分子、金属离子等。
参考文献:Analysis of CDK2 Active-SiteHydration: A Method to Design New Inhibitors Zdeneˇk Krˇ?′zPROTEINS: Structure, Function, and Bioinformatics 55:258–274 (XX)12.2 分子对接①聚合物对接前效果图②聚合物对接后效果图对接后实际距离和设置的最优值12-3-2在样本文件中,创建冰的晶体结构,分别做温度为260K,273K,298K,373K下的分子动力学模拟(10 ps),观察晶体机构的变化情况,并做定性解释。
①不同温度下冰晶体结构图:原始冰晶体结构图由冰晶体在不同温度下的结构可见,随温度升高,冰晶体的各个水分子之间的距离不断增加,晶体结构趋向于分散无序状。
②不同温度下,冰晶体分子动力学模拟图③不同温度下体系的总能量与势能由曲线形状可见,经过分子动力学模拟之后,体系的能量降低,变得更加稳定。
由计算结果可见,体系的总能量和势能随温度的升高而增大。
因为当温度升高时,分子的热运动加剧,使分子的伸缩、转动、振动势能增加从而使分子总能量增加,而体系的是能增加是因为非键相互作用尤其是分子间氢键相互作用减弱。
二、实验心得与体会本次实验主要进行了生物大分子的模拟。
生物大分子一般包含上千个原子,目前还不能应用量子化学从头计算方法模拟,常用的方法有QM/MM方法,和纯粹的分子动力学模型。
1.关于分子力学要求掌握四点内容:(1)分子力学中,离子间的相互作用势能函数是什么?(2)势函数中存在特定的参数,怎么给参数赋初值?(3)原子类型怎样确定?(4)力场有哪些?各自的适用范围是什么?下面详细解释:(1)V(r)有四项,前三项对应于键伸缩势能、弯曲势能和扭转势能。

一、实验目的1. 理解分子荧光光谱分析的基本原理和操作方法;2. 掌握荧光光谱仪器的组成及各部分作用;3. 分析影响荧光强度的内部结构因素和外部环境因素;4. 了解光谱分析法的应用范围。
二、实验原理分子荧光光谱分析是利用某些物质分子受光照射时所发生的荧光的特性和强度,进行物质的定性分析或定量分析的方法。
当分子吸收紫外和可见光后,电子跃迁到激发态,随后以发射辐射的方式释放能量,再回到基态。
如果发射的波长与吸收的波长相同或不同,这种现象称为光致发光,其中最常见的光致发光现象是荧光和磷光。
荧光光谱分析主要包括激发光谱、发射光谱、同步光谱和三维荧光光谱。
激发光谱表示激发光波长与荧光强度之间的关系,发射光谱表示荧光光波长与荧光强度之间的关系。
同步光谱是指激发光波长和发射光波长同时改变时,荧光强度的变化情况。
三维荧光光谱是指在三维坐标系中,激发光波长、发射光波长和荧光强度之间的关系。
影响荧光强度的因素包括内部结构因素和外部环境因素。
内部结构因素主要包括分子的共轭程度、取代基、分子结构等。
外部环境因素主要包括溶剂、温度、pH值、浓度等。
三、实验内容与步骤1. 实验仪器与试剂:荧光光谱仪、激发光源、样品池、标准样品、溶剂等。
2. 实验步骤:(1)将荧光光谱仪开机预热,调整好仪器参数;(2)将标准样品放入样品池,调整样品池位置;(3)设置激发光波长,进行激发光谱扫描;(4)设置发射光波长,进行发射光谱扫描;(5)设置同步光谱参数,进行同步光谱扫描;(6)设置三维荧光光谱参数,进行三维荧光光谱扫描;(7)记录实验数据,分析数据,得出结论。
四、实验结果与分析1. 激发光谱扫描结果显示,标准样品在特定波长范围内有明显的荧光峰,说明该样品在该波长范围内具有荧光特性。
2. 发射光谱扫描结果显示,标准样品在激发光波长下具有明显的发射峰,说明该样品在该激发光波长下具有荧光发射特性。
3. 同步光谱扫描结果显示,激发光波长和发射光波长同时改变时,荧光强度也随之变化,说明激发光波长和发射光波长对荧光强度有显著影响。
分子模型实验报告篇一:分子模拟实验实验报告生物大分子分子模拟实验作业——生物大分子一、实验部分12-3-1获得PDB号为“1HCK”的蛋白(human-cyclin-dependent kinase 2,i,e.,CKD2和ATP的结合晶体结构),并采用不同的模型观察其特点①分别用卡通模型和丝带模型显示生物大分子结构,并用球棍模型、棒状模型显示其中小分子、金属离子等。
参考文献:Analysis of CDK2 Active-SiteHydration: A Method to Design New Inhibitors Zdeneˇk Krˇ?′zPROTEINS: Structure, Function, and Bioinformatics 55:258–274 (XX)12.2 分子对接①聚合物对接前效果图②聚合物对接后效果图对接后实际距离和设置的最优值12-3-2在样本文件中,创建冰的晶体结构,分别做温度为260K,273K,298K,373K下的分子动力学模拟(10 ps),观察晶体机构的变化情况,并做定性解释。
①不同温度下冰晶体结构图:原始冰晶体结构图由冰晶体在不同温度下的结构可见,随温度升高,冰晶体的各个水分子之间的距离不断增加,晶体结构趋向于分散无序状。
②不同温度下,冰晶体分子动力学模拟图③不同温度下体系的总能量与势能由曲线形状可见,经过分子动力学模拟之后,体系的能量降低,变得更加稳定。
由计算结果可见,体系的总能量和势能随温度的升高而增大。
因为当温度升高时,分子的热运动加剧,使分子的伸缩、转动、振动势能增加从而使分子总能量增加,而体系的是能增加是因为非键相互作用尤其是分子间氢键相互作用减弱。
二、实验心得与体会本次实验主要进行了生物大分子的模拟。
生物大分子一般包含上千个原子,目前还不能应用量子化学从头计算方法模拟,常用的方法有QM/MM方法,和纯粹的分子动力学模型。
1.关于分子力学要求掌握四点内容:(1)分子力学中,离子间的相互作用势能函数是什么?(2)势函数中存在特定的参数,怎么给参数赋初值?(3)原子类型怎样确定?(4)力场有哪些?各自的适用范围是什么?下面详细解释:(1)V(r)有四项,前三项对应于键伸缩势能、弯曲势能和扭转势能。
分子光谱实验报告分子光谱实验报告引言:分子光谱实验是一种重要的实验方法,它通过研究分子在不同波长的光照射下的吸收、发射或散射现象,来揭示分子的结构和性质。
本次实验旨在通过红外光谱和紫外-可见光谱两种光谱方法,对不同分子进行分析与探究。
实验一:红外光谱分析红外光谱分析是一种研究分子结构的重要手段。
在该实验中,我们选取了苯酚、乙醇和甲醇作为实验样品,利用红外光谱仪测量它们在不同波长下的吸收情况。
结果显示,苯酚在红外光谱中出现了两个宽而强烈的吸收峰,分别位于3400cm-1和1600 cm-1附近。
这表明苯酚分子中存在有羟基(-OH)和芳香环基团。
乙醇的红外光谱中也出现了一个宽而强烈的吸收峰,位于3400 cm-1附近,这表明乙醇分子中存在有羟基(-OH)。
而甲醇的红外光谱中则只出现了一个宽而强烈的吸收峰,位于3400 cm-1附近,这表明甲醇分子中也存在有羟基(-OH)。
通过对红外光谱的分析,我们可以确定分子中存在的官能团,进而推测分子的结构和性质。
实验二:紫外-可见光谱分析紫外-可见光谱分析是一种研究分子电子结构和电子能级的方法。
在该实验中,我们选取了苯酚、乙醇和甲醇作为实验样品,利用紫外-可见光谱仪测量它们在不同波长下的吸收情况。
结果显示,苯酚在紫外-可见光谱中出现了一个吸收峰,位于270 nm附近。
乙醇的紫外-可见光谱中也出现了一个吸收峰,位于210 nm附近。
而甲醇的紫外-可见光谱中则出现了两个吸收峰,分别位于190 nm和270 nm附近。
通过对紫外-可见光谱的分析,我们可以了解分子中的电子能级分布情况,进而推测分子的电子结构和性质。
实验三:红外光谱和紫外-可见光谱的对比分析在实验一和实验二的基础上,我们对苯酚、乙醇和甲醇的红外光谱和紫外-可见光谱进行了对比分析。
结果显示,苯酚在红外光谱和紫外-可见光谱中的吸收峰位置并无明显的对应关系。
这说明红外光谱和紫外-可见光谱能够从不同角度揭示分子的性质和结构。
分⼦模拟实验综合实验分⼦模拟实验作业——综合实验⼀、实验部分1.反应物和产物分⼦的电⼦结构(1)画出反应物和产物分⼦的⽴体结构图,分别⽤“wireframe(线条型)”,“ball & stick(球棍型)”,“cylindrical bonds(键线型)”,“space filing(实⼼球型)”(3)计算分⼦轨道,并图⽰各个分⼦的HOMO和LUMO轨道的形状和能量。
将所有的分⼦轨道按能级排列次序,并以此分析两反应物的轨道匹配情况。
C4H6LUMO HOMOC2H4LUMO HOMOC6H10LUMO HOMO反应是由1,3-丁⼆烯的HOMO与⼄烯的LUMO反应,故图为分⼦轨道很匹配,有利于加成反应。
DA反应的反应物分为两部分,双烯体提供共轭双烯,亲双烯体提供不饱和键。
DA反应是由双烯体的HOMO与亲双烯体的LUMO发⽣作⽤,反应过程中,电⼦从双烯体的HOMO流⼊亲双烯体的LUMO。
C2H4的LUMO轨道与C4H6的HOMO轨道电⼦云相匹配,正好符合双烯合成的条件,利于反应的进⾏。
(4)绘制反应物和产物分⼦的总电⼦密度图和静电势图,分析两个反应物分⼦的电性匹配情况。
总电⼦密度图静电势图总电⼦密度图静电势图由轨道匹配来说,要使得反应能够顺利进⾏,必须是同为正或负。
C2H4的静电势图与C4H6的静电势,很匹配,所以有利于加成反应。
2.构象搜索与分⼦间长程相互作⽤(1)计算丁⼆烯分⼦绕CCCC⼆⾯⾓转动的构象,确定稳定构型,并计算内转动的能垒⾼度。
(可能不⽌⼀个)此时的构型有两种,顺式构型和反式构型。
稳定构型为反式构型,内转动能垒⾼度:40.58kcal/mol(反式构型)和38.67kcal/mol(顺式构型)。
图1 扫描势能⾯时的构型能量单位:kcal/mol①势能⾯图:图2丁⼆烯分⼦绕CCCC ⼆⾯⾓转动的构象②相互作⽤势能线及拟合图选取每个距离下的最低能量,减去MM2优化得到的C2H4和C4H6的能量 C2H4:Total: 0.4267 C4H6:Total: -0.1615 数据表格如下:CCCC ⼆⾯⾓/(°)质⼼距离/?质⼼距离/?相互作⽤能量(kc al/图3 C 4H 6与C 2H 4相互作⽤势能曲线3 反应途径计算(1)在HF/6-31G(d)理论⽔平上计算两个反应物分⼦和产物分⼦的298.15K时标准⽣成焓。
武汉大学化学与分子科学学院《分子模拟实验》实验报告设计实验——有关消去反应的探究指导老师:侯华姓名:陆文心专业:化学弘毅班学号:2012301040179日期:2015年1月8日(周四下午)一、背景介绍消去反应(elimination )是有机化学中一类常见的反应,根据机理的不同,主要可分为E1(单分子消去反应)、E2(双分子消去反应)和E1cb (单分子共轭碱消去反应)等类型。
它作为一类常见而常用的反应,其机理很早就被研究出来。
本实验旨在从计算化学的角度,利用已掌握的Chem3D 软件功能,结合本学期所学的一些分子模拟实验相关知识,对一个典型的消去反应:CH 3CH 2Br ——→OH -CH 2=CH 2 + HBr作出相关探究。
二、主要问题(1)资料显示,该反应属于E2反应。
设计出此反应的过渡态,并通过比较过渡态与原始构型,说明该反应的过程。
(2)绘出此反应的反应途径,计算活化能及反应热。
(3)探究溶剂(水、乙醇、甲苯)对此反应的影响。
(4)作出反应物CH 3CH 2Br 的红外及拉曼光谱,比较并分析二者的异同。
三、实验方案1. 过渡态的优化根据反应机理,氢氧根离子进攻与溴原子相连的碳原子邻位的碳原子,同时保证与溴原子处于“反式共平面”的位置关系。
由于该反应为双分子消去反应,氢原子与溴原子的离去是同时发生的,故可设计出反应的过渡态。
优化方法选用最常用的HF,关键在于基组的选择。
常用的为6-31,由于此反应物中含有重原子Br,为保证计算结果的准确性,使用弥散函数sp;由本学期所学的知识,在不确定壳层性质的情况下,优先选用RO(Restricted Open-Shell)。
故选用理论水平为ROHF/6-31++G(d)。
过渡态中氢氧根离子所带电荷为-1,故过渡态的净电荷为-1。
综上所述,相关设置界面如下:优化完毕,应计算频率,若只有一个虚频率,才可判断此过渡态为真正的过渡态。
2. 反应途径的绘制与热力学量的计算由Optimize to Transition State可得出过渡态的能量。
分子模拟实验分子光谱模拟实验内容介绍:分子光谱是化学研究的重要组成之一,它是联系物质的微观结构和宏观性质的桥梁,各种光谱技术已经成了表征化合物结构的必要手段。
原则上讲,实验室观察到的光谱是大量分子在特定环境和特定激发条件下的统计表现,无法或很难从理论的角度去模拟实验光谱。
但是任何一种光谱都有其深刻的理论原理,比如紫外光谱是与电子激发态密切关联的,激发态的性质、能级、跃迁强度等都可以反映为光谱。
虽然量子化学无法计算得到实际的光谱,但可以模拟更本质的东西,也可以与实验光谱的某些特征(比如谱峰位置)相比较,同时有助于实验光谱峰的确认。
本实验就最常见的几种分子光谱作详细的介绍,通过分子模拟计算来说明量子化学是如何模拟光谱的。
主要的实验内容包括紫外可见光谱、拉曼光谱和核磁共振(NMR)谱。
实验要求:1、掌握各种常见光谱图的量子化学计算方法;2、掌握分析光谱的技巧;3、了解理论光谱图的价值和意义;4、学会将理论知识结合实验谱图作出分析比较。
实验一:紫外可见光谱的模拟紫外可见光谱是分子中电子被激发到高激发态时,在不同电子能级之间跃迁所形成的吸收(从低能级到高能级)或荧光(从高能级到低能级)光谱。
用GAMESS 程序的多参考态自洽场(MCSCF)中的单组态相互作用方法(CIS)。
通过CIS 方法计算电子激发态,估算紫外可见光谱中吸收峰的位置。
计算HCOOH分子五个垂直激发的单重态和三重态,2个绝热激发的单重态和3个三重态,从而确定垂直激发和绝热激发的波长。
采用的优化方法和基组为HF/6-31G(d),得到的数据为下表所示。
1、垂直激发表一:5个垂直激发的单重态能量数据表二:5个垂直激发的三重态能量数据转化为激发波长,采用的公式为1nm=(107/∆E)cm-1,∆E为CM-1一栏的能量,结果如下表:2、绝热激发转化为激发波长,采用的公式为1nm={107(/∆E×349.755)}cm-1,∆E为单位为KCAL.MOL-1的能量,结果如下表:小结:1、垂直激发中激发态构型与基本构型相同;2、绝热激发态中构型发生比较明显的改变;3、可见垂直激发态中,三重态的波长总体大于单重态,因此垂直激发时,单重态的能量要高于三重态,因此单重态中两个单电子的自旋方向相反,而三重态中的两个单电子自旋相同;4、绝热激发态的波长总体上大于垂直激发的波长,其单重态的波长小于三重态;5、两种激发类型共同存在的趋势:激发态的数目越大,波长越小;通常意义上的光谱对应于垂直激发。
分子模拟实验分子光谱模拟实验内容介绍:分子光谱是化学研究的重要组成之一,它是联系物质的微观结构和宏观性质的桥梁,各种光谱技术已经成了表征化合物结构的必要手段。
原则上讲,实验室观察到的光谱是大量分子在特定环境和特定激发条件下的统计表现,无法或很难从理论的角度去模拟实验光谱。
但是任何一种光谱都有其深刻的理论原理,比如紫外光谱是与电子激发态密切关联的,激发态的性质、能级、跃迁强度等都可以反映为光谱。
虽然量子化学无法计算得到实际的光谱,但可以模拟更本质的东西,也可以与实验光谱的某些特征(比如谱峰位置)相比较,同时有助于实验光谱峰的确认。
本实验就最常见的几种分子光谱作详细的介绍,通过分子模拟计算来说明量子化学是如何模拟光谱的。
主要的实验内容包括紫外可见光谱、拉曼光谱和核磁共振(NMR)谱。
实验要求:1、掌握各种常见光谱图的量子化学计算方法;2、掌握分析光谱的技巧;3、了解理论光谱图的价值和意义;4、学会将理论知识结合实验谱图作出分析比较。
实验一:紫外可见光谱的模拟紫外可见光谱是分子中电子被激发到高激发态时,在不同电子能级之间跃迁所形成的吸收(从低能级到高能级)或荧光(从高能级到低能级)光谱。
用GAMESS程序的多参考态自洽场(MCSCF)中的单组态相互作用方法(CIS)。
通过CIS方法计算电子激发态,估算紫外可见光谱中吸收峰的位置。
计算HCOOH分子五个垂直激发的单重态和三重态,2个绝热激发的单重态和3个三重态,从而确定垂直激发和绝热激发的波长。
采用的优化方法和基组为HF/6-31G(d),得到的数据为下表所示。
1、垂直激发表一:5个垂直激发的单重态能量数据表二:5个垂直激发的三重态能量数据转化为激发波长,采用的公式为1nm=(107/?E)cm-1,?E为CM-1一栏的能量,结果如下表:表三:HF/6-31G(d)理论水平下HCOOH分子的垂直激发波长表(单位:nm)1S 2S 3S 4S 5S激发波长单位:nm 179 120 111 110 100 1T 2T 3T 4T 5T198 195 122 116 1092、绝热激发表三:绝热激发态下甲酸模型和参数HCOOH分子状态模型参数和能量基态RC1-O2=1.18ARC1-H4=1.08ARC1-03=1.32ARO3-H5=0.95A∠O2-C1-O3=124.94°∠H4-C1-03=110.45°E= -118450.038151kcal/mol第一个单重绝热激发态(111)RC1-O2=1.28ARC1-H4=1.08ARC1-03=1.37ARO3-H5=0.95A∠O2-C1-O3=115.17°∠H4-C1-03=113.50°E= -118309.60242kcal/mol E= 140.44 kcal/mol第一个单重绝热激发态(212)RC1-O2=1.46ARC1-H4=1.09ARC1-03=1.30ARO3-H5=0.95A∠O2-C1-O3=113.18°∠H4-C1-03=110.66°E= -118260.935089kcal/mol E= 189.10 kcal/mol第一个三重绝热激发态(131)RC1-O2=1.39ARC1-H4=1.07ARC1-03=1.36ARO3-H5=0.95A∠O2-C1-O3=112.65°∠H4-C1-03=110.92°E= -118346.920346kcal/mol E= 103.12 kcal/mol第二个三重绝热激发态(232)RC1-O2=1.29ARC1-H4=1.08ARC1-03=1.38ARO3-H5=0.95A∠O2-C1-O3=112.35°∠H4-C1-03=110.83°E= -118329.140018 kcal/molE= 120.90 kcal/mol第三个三重绝热激发态(333)RC1-O2=1.45ARC1-H4=1.09ARC1-03=1.31ARO3-H5=0.95A∠O2-C1-O3=112.71°∠H4-C1-03=113.38°E= -118270.928983 kcal/molE= 179.11 kcal/mol转化为激发波长,采用的公式为1nm={107/(?E×349.755)}cm-1,?E为单位为KCAL.MOL-1的能量,结果如下表:表四:绝热激发态下激发波长1S 2S激发波长单位(nm)204 1511T 2T 3T 277 237 160小结:1、垂直激发中激发态构型与基本构型相同;2、绝热激发态中构型发生比较明显的改变;3、可见垂直激发态中,三重态的波长总体大于单重态,因此垂直激发时,单重态的能量要高于三重态,因此单重态中两个单电子的自旋方向相反,而三重态中的两个单电子自旋相同;4、绝热激发态的波长总体上大于垂直激发的波长,其单重态的波长小于三重态;5、两种激发类型共同存在的趋势:激发态的数目越大,波长越小;通常意义上的光谱对应于垂直激发。
实验名称:分子光谱分析实验日期:2023年3月15日实验地点:化学实验室实验人员:张三、李四、王五一、实验目的1. 熟悉分子光谱仪器的操作方法。
2. 掌握分子光谱的基本原理和应用。
3. 分析样品的分子光谱,确定其组成和结构。
二、实验原理分子光谱是指分子在吸收或发射光的过程中,产生的特定波长的光谱。
分子光谱可分为紫外-可见光谱、红外光谱、拉曼光谱等。
本实验采用紫外-可见光谱仪对样品进行检测。
紫外-可见光谱的原理是基于分子吸收特定波长的光子,使分子内部的电子发生跃迁。
通过分析吸收光谱,可以确定样品的组成和结构。
三、实验仪器与试剂1. 仪器:紫外-可见光谱仪、分析天平、移液器、容量瓶、试管等。
2. 试剂:待测样品、溶剂、标准溶液等。
四、实验步骤1. 准备样品:称取一定量的待测样品,用溶剂溶解,配制成一定浓度的溶液。
2. 标准曲线绘制:配制一系列标准溶液,分别测定其在特定波长下的吸光度,以吸光度为纵坐标,浓度为横坐标,绘制标准曲线。
3. 样品测定:将样品溶液注入紫外-可见光谱仪,在特定波长下测定其吸光度。
4. 数据处理:根据标准曲线,计算样品的浓度。
五、实验结果与分析1. 标准曲线绘制根据实验数据,绘制标准曲线如下:```吸光度(y)浓度(x)0.1 100.2 200.3 300.4 400.5 50```2. 样品测定根据实验数据,样品溶液在特定波长下的吸光度为0.35。
3. 数据处理根据标准曲线,计算样品的浓度为:浓度 = 0.35 × (50 - 10) / (0.5 - 0.1) = 30 mg/L六、实验结论通过本次实验,我们成功掌握了分子光谱仪器的操作方法,并学会了如何分析样品的分子光谱。
实验结果表明,样品在特定波长下的吸光度与其浓度呈线性关系,从而确定了样品的浓度。
七、实验讨论1. 实验过程中,应注意样品的纯度和溶液的浓度,以确保实验结果的准确性。
2. 在绘制标准曲线时,应选择合适的浓度范围,避免出现线性关系不明显的情况。
1.红外光谱:分别用 PM3 ,HF/6-31G(d),B3LYP/6-31G(d),MP2/6-31G(d) 四种理论方法计算 H 2O 分子的红外光谱,并比较结果的优劣。
实验 上测得的水分子的振动频率为:1594cm 1, 3657cm -1, 3756cm 1。
k(Lore ntz)
B3LYP/6-31G(d)
由图像可得四种理论方法得到的振动频率分别为
PM3 1698 cm -1、3770 cm -1、3880 cm -1 HF/6-31G(d) 1634 cm -1、3662 cm -1、3770 cm -1 B3LYP/6-31G(d)
1634 cm -1、3566 cm -1、3662 cm -1 MP2/6-31G(d)
1644 cm -1、3544 cm -1、 3684 cm -1
与标准值1594cm -1, 3657cm'1,3756cm'1比较,HF/6-31G(d)最为接近标准值;
PM3三个频率都偏大,与标准值符合情况不好; B3LYP/6-31G(d)除1634 cm -1与
分子模拟实验作业 一、实验部分
分子光谱模拟
k(Lore ntz)
-DrnelPL (.
4000
3000
2000
-1
/cm
1000
'!
0.00
0.01
0.02
0.03
0.04
0.05
0.06
4000
3000
2000 1000
/cm
-1
PM3
HF/6-31G(d)
------ k(Lore ntz)
k(Lore ntz)
0.00
0.01 r
0.04
0.05 J _______ ! ______ ______ J ______ ______ !_
4000 3000 2000
-1
/cm
1000
MP2/6-31G(d)
0.000
0.002 _
0.004
0.006
0.008
0.010
0.012 - 0.014
0.02
0.03 -
e
/cm
标准值较接近外,其余两个频率均偏小;MP2/6-31G(d) 1644 cm-1与标准值接近,
其余两个频率均偏小
2•拉曼光谱的模拟
HF/6-31G(d)计算的CH4分子的拉曼谱图
111
图中特征波数为3290 cm、3189 cm、1705 cm
3•紫外可见光谱的模拟
计算甲酸分子5个垂直激发的单重态和三重态,2个绝热激发的单重态和三重态,并确定垂直激发和绝热激发波长。
(1)垂直激发
CI-SINGLES EXCITATION ENERGIES
STATE HARTREE EV KCAL/MOL CM-1
1A 0.2550545872 6.9404 160.0492 55978.01
1A 0.3796011178 10.3295 238.2033 83312.82
1A 0.4095383929 11.1441 256.9893 89883.29
1A 0.4158054412 11.3146 260.9219 91258.75
1A 0.4554734351 12.3941 285.8139 99964.86
HF/6-31G(d)理论水平下HCOOH分子5个单重激发态的垂直激发能
CI-SINGLES EXCITATION ENERGIES
STATE HARTREE EV KCAL/MOL CM-1
3A 0.2299108089 6.2562 144.2712 50459.59 3A 0.2330765536 6.3423 146.2578 51154.39 3A 0.3720691489 10.1245 233.4769 81659.74 3A 0.3918314869 10.6623 245.8780 85997.07 3A 0.4193773712 11.4118 263.1633 92042.69
HF/6-31G(d)理论水平下HCOOH分子5个三重激发态的垂直激发能
HF/6-31G(d)理论水平下HCOOH分子垂直激发波长表(单位:nm)
(2)绝热激发
E=-118309.6kcal/mol 第一个单重激发态
E=-11826O.9 kcal/mol
第二个单重激发态
△ E=189.1 kcal/mol
E=-118346.9kcal/mol
第一个三重激发态
△ E=103.1 kcal/mol
E=-118309.6 kcal/mol
第二个三重激发态
△ E=103.1 kcal/mol
△ E=140.4 kcal/mol
可见,垂直激发态中,三重态的波长总体看来大于单重态,因此垂直激发时, 单重态的能量要高于三重态的能量,因为单重态中两个单电子的自旋方向相反,
而三重态中两个单电子自旋相同。
绝热激发态的波长总体上大于垂直激发的波长,其余规律与垂直激发类似。
两种激发类型共同存在的趋势:激发态的数目越大,波长越小。
4.核磁共振(NMR )谱的模拟
AS H2)=(31.2+30.3)/2-33.4= -2.65
AS HN)=(32.4+31.9)/2-33.4= -1.25
AS (0)=27.8-33.4= -5.6
AS CH2)=200.2-257.2= -57
AS C=O)=100.7-257.2= -156.5
NMR的结果显然HF/STO-3G理论水平的计算值不可靠。