刘泉鹏讲解运动神经元科普知识
- 格式:doc
- 大小:5.69 KB
- 文档页数:3





【每日一讲】第329期α、γ运动神经元与疼痛 郑大解剖学 腾康学院 每日一讲 天天有进步 每天07:00开始
第329期 α、γ运动神经元与疼痛 2020/11/25
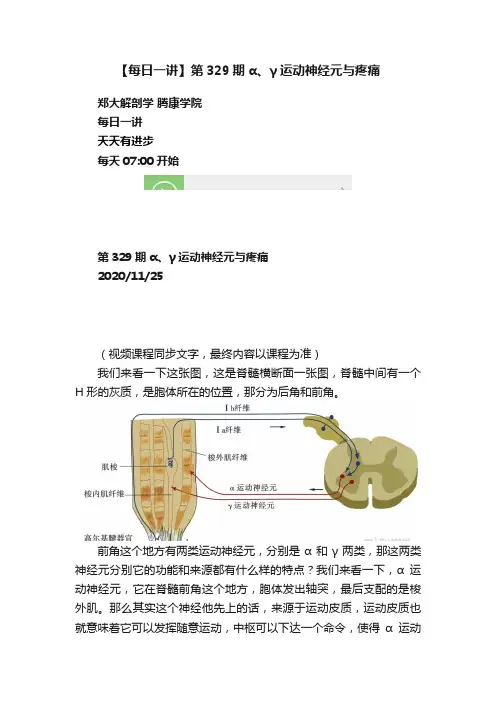
(视频课程同步文字,最终内容以课程为准) 我们来看一下这张图,这是脊髓横断面一张图,脊髓中间有一个H形的灰质,是胞体所在的位置,那分为后角和前角。
前角这个地方有两类运动神经元,分别是α和γ两类,那这两类神经元分别它的功能和来源都有什么样的特点?我们来看一下,α运动神经元,它在脊髓前角这个地方,胞体发出轴突,最后支配的是梭外肌。那么其实这个神经他先上的话,来源于运动皮质,运动皮质也就意味着它可以发挥随意运动,中枢可以下达一个命令,使得α运动神经元兴奋,梭外肌收缩。 那么再有α运动神经元它支配的肌肉是梭外肌,这个梭外肌属于典型肌纤维,也就是我们所说的肌纤维,大部分就指的是梭外肌。那么一个运动神经元,α运动神经元所支配的这个梭外肌叫做运动单元。
因为一个胞体发出的轴突,这个突起支配不止一条这个肌纤维,是多个。那么不同部位这些数量还不太一样。比如手部的肌肉蚓状肌,一个胞体支配三条肌纤维,但是小腿这个地方腓肠肌,一个胞体最后支配1600这个肌纤维。
α运动神经元在结合这个梭外肌,再加上它这种中枢的联系,最后可以发挥姿势运动平衡,包括习惯活动中的随意运动,还包括非随意运动。那虽然它来源于大脑的运动皮质,当然有一部分是皮质下,所以也发挥非随意肌纤维的这个收缩,发出这样的神经通冲动。 那么第二个是γ运动神经元,它在脊髓前角这个地方胞体发出轴突。那往上走呢?它的这个中枢来源于脑干。那么胞体发出的轴突,最后支配的肌纤维和α运动神经元不一样,它支配的是梭内肌。那梭内肌收缩,可以影响肌梭的敏感性,一收缩可以使它的敏感性增加。
那么由于它向上,来源的信息是在脑干里面,所以是无意识的,这些信息从脑干这个地方发出,接着使得γ运动神经元有一定的神经张力,来设定肌梭的敏感性,从而设定肌张力。那其实最终是通过随意肌,也就是梭外肌收缩来负责有意识的精细肌肉的控制。 那所以α和γ运动神经元一般是同时激活,这个是它所对应的肌肉收缩,激活主动肌、拮抗肌、协同肌,来保证动作的协调和灵活性,这是功能。 损伤,如果组织损伤,炎症等,引起了化学刺激或者是肿胀,最后造成骨骼肌这个地方的缺血,损伤而引起疼痛,而这种疼痛和肿胀可以造成感受器功能的障碍,可以造成比如肌梭感受器功能的障碍,那接着会造成这个肌肉本体感觉能力下降,平衡能力下降,姿势异常,反应慢,精细运动的调节能力也会是出现下降,那么肌肉激活的这个肌肉激活的顺序也会发生改变,这是疼痛和肿胀以后会造成的一个结果。 那么第二种,如果是慢性,比如这个人曾经有过外伤,接着出现了粘连,筋膜的粘连或者是不平衡老化,全身整体情况下降,比如年龄增加了,全身退行性病变,造成慢性肌肉功能障碍。


运动神经元治好案例
在运动神经元治疗方面,有一个令人振奋的案例,让我们一起来了解一下。
这个案例中,患者是一位年轻的男性,因为运动神经元疾病而备受折磨。
他曾
经是一名优秀的运动员,但是随着疾病的加重,他的身体逐渐失去了力量和灵活性。
他的生活变得越来越困难,无法自理,甚至连简单的日常活动也变得困难重重。
在经历了多次治疗无效之后,他找到了一位运动神经元专家。
这位专家经过仔
细的诊断和分析,制定了一套针对性的治疗方案。
这个方案包括药物治疗、物理疗法和营养调理等多个方面,综合治疗,旨在从根本上改善患者的病情。
在经过一段时间的治疗之后,患者的病情出现了积极的变化。
他的肌肉逐渐恢
复了力量,身体的灵活性也有了明显的改善。
更重要的是,他的生活质量得到了极大的提高,重新找回了生活的乐趣和活力。
这个案例告诉我们,运动神经元疾病并非绝症,只要找对方法,采取正确的治
疗措施,就有可能得到有效的改善。
同时,也提醒我们,对于这类疾病,及时就医,找到专业的医生和医疗机构进行治疗至关重要。
不要因为疾病而放弃治疗,更不要盲目跟风尝试各种不明确的疗法。
总之,这个案例为我们提供了一个积极的启示,运动神经元疾病是可以治愈的,只要我们积极应对,找到专业的医疗机构和医生,采取科学的治疗方案,就有可能战胜疾病,重拾健康和活力。
希望这个案例能给正在经历类似疾病的患者带来信心和希望,也希望更多的人能够关注和重视这类疾病的治疗和研究,为患者带来更多的福音。


运动神经元症引言:运动神经元症(Motor Neuron Disease,简称MND)是一类神经系统疾病,主要影响人体的神经元,包括脊髓运动神经元和脑神经元,进而影响肌肉活动。
该疾病的发病率虽然相对较低,但却给患者、家庭和医疗团队带来了极大的困扰。
本文将对运动神经元症的病因、症状、诊断和治疗进行详细的介绍,以便更好地了解和对抗这一疾病。
一、病因1. 基因突变:在某些运动神经元症患者身上,研究者发现了一些突变的基因,这些基因与神经元的功能和生存有关。
2. 环境因素:尽管没有确凿的证据证明,但一些环境因素,如毒物暴露和慢性创伤,也与运动神经元症的发病率有所关联。
二、症状1. 肌肉无力:运动神经元症的患者往往会出现肌肉无力的症状,通常从一侧肢体开始,逐渐发展到其他部位。
2. 肌肉抽搐:患者可能会在肌肉中出现不自主的抽搐,特别是在休息时更加明显。
3. 智力下降:在某些运动神经元症的亚型中,患者会出现认知功能的下降,甚至出现失语和思维迟缓等症状。
4. 嚼食和吞咽困难:由于运动神经元症会影响颌面肌肉的活动,患者会出现嚼食和吞咽困难的情况。
三、诊断1. 体格检查:医生会通过观察病人的肌肉活动和运动能力来进行初步的诊断。
2. 电生理检查:通过电生理检查,医生可以观察到患者神经传导的异常情况,帮助进一步确定是否为运动神经元症。
3. 化验:化验可以帮助排除其他疾病的可能,并通过检查血液和尿液中的生化指标来评估患者的肝肾功能。
四、治疗1. 药物治疗:目前,尚无特效药物可以治愈运动神经元症,但一些药物可以用于缓解症状,如肌肉抽搐和运动障碍。
2. 物理治疗:物理治疗师可以通过特定的运动和手法来帮助患者改善肌肉活动和运动功能。
3. 呼吸支持:当病情进展到影响呼吸肌肉时,患者可能需要呼吸支持设备,如呼吸机或胸部外固定器。
4. 康复护理:康复护理团队可以为患者提供全面的支持,帮助他们应对身体和心理上的挑战。
结论:虽然运动神经元症目前还没有治愈的方法,但通过及时的诊断和综合的治疗措施,可以缓解患者的症状,提高他们的生活质量。
运动神经元病(MND)是一组病因未明的选择性侵犯脊髓前角细胞、脑干运动神经元、皮层锥体细胞及锥体束的慢性进行性神经变性疾病。
发病率约为每年1~3/10万,患病率为每年4~8/10万。
由于多数患者于出现症状后3~5年内死亡,因此,该病的患病率与发病率较为接近。
MND病因尚不清楚,一般认为是随着年龄增长,由遗传易感个体暴露于不利环境所造成的,即遗传因素和环境因素共同导致了运动神经元病的发生。
根据大量流行病学调查,人们发现了许多与ALS发病相关的环境因素,包括重金属、杀虫剂、除草剂、外伤、饮食以及运动等。
但是总体来讲,这些因素之间缺乏联系,而且它们与ALS的发生是否存在必然联系以及它们导致ALS发生的机制也有待进一步证实
运动神经元病的疾病类型
运动神经元病包括肌萎缩侧索硬化、进行性脊肌萎缩症、原发性侧索硬化和进行性延髓麻痹。
各种类型的运动神经元疾病的病变过程大都是相同的,主要差别在于病变部位的不同。
可将肌萎缩侧索硬化症看作是本组疾病的代表,其它类型则为其变型。
肌萎缩侧索硬化(amyotrophic lateral sclerosis,ALS)在早先时期与运动神经元疾病具有完全等同的含义,特指先有下运动神经元损害,之后又有上运动神经元损害的一个独立的疾病。
但后来发现还有另外两种变异情况,即病程中始终只累及上运动神经元或下运动神经元,前者称为原发性侧索硬化,后者称为脊髓性肌萎缩。
但有些文献仍沿用运动神经元病来专指肌萎缩侧索硬化。
多数学者习惯根据上、下运动神经元受累的不同组合,将运动神经元病分为肌萎缩侧索硬化、原发性侧索硬化和脊髓性肌萎缩三种类型。
肌萎缩侧索硬化与多种相关疾病有共同的病理基础,这些疾病包括原发性侧索硬化、ALS-痴呆、ALS-相关性额叶痴呆、进行性脊髓性肌萎缩、多系统萎缩和lewy小体病。
病理检查发现这些疾病同样含有泛素阳性包涵体和透明团块包涵体,只是损伤了不同的解剖部位而出现各种各样的临床组合。
运动神经元病的发病机制
确切的发病机制至今尚未清楚。
研究主要集中在铜锌超氧歧化酶基因突变学说、兴奋性氨基酸毒性学说、自身免疫学说和神经营养因子学说。
1.铜锌超氧歧化酶基因突变学说
研究表明,20%的家族性ALS有SODI(Cu/Zn过氧化物歧化酶)基因突变。
该基因位于人类染色体21q22.1,其突变可致SODl活性丧失,使超氧化的解毒作用减弱,致自由基过量积聚,细胞损伤。
一些散发性的ALS可能也存在2lq22位点的突变。
2.兴奋性氨基酸毒性学说
兴奋性氨基酸包括谷氨酸、天冬氨酸及其衍生物红藻氨酸(KA)、使君子氨酸(QA)、鹅膏氨酸(IA)和N-甲基d-天冬氨酸(NMDA)。
兴奋性氨基酸的兴奋毒性可能参与LIS的发病。
谷氨酸与NMDA受体结合可致钙内流,激活一系列蛋白酶和蛋白激酶,使蛋白质的分解和自由基的生成增加,脂质过氧化过程加强,神经元自行溶解。
此外过量钙还可激活核内切酶,使DNA
裂解及核崩解。
ALS的病变主要局限在运动神经系统可能与谷氨酸的摄取系统有关。
这个摄取系统位于神经胶质细胞及神经细胞的细胞膜,能迅速将突触间隙的谷氨酸转运到细胞内,终止其作用。
研究发现ALS的皮质运动细胞、脊髓胶质细胞和脊髓灰质细胞的谷氨酸摄取系统减少。
动物实验研究也显示小鼠鞘内注射KA及NMDA可致脊髓神经元变性。
3.自身免疫学说
ALS患者脑脊液和血清中抗神经元抗体的增加提示其发病可能与自身免疫有关。
如存在于ALS患者血清中的L型电压依赖性钙通道抗体可与该通道蛋白结合,改变其电生理特性,造成神经元损伤。
4.病理改变
显微镜下观察可见脊髓前角细胞减少,伴胶质细胞增生,残存的前角细胞萎缩。
大脑皮质的分层结构完整,锥体细胞减少伴胶质细胞增生。
脊髓锥体束有脱髓鞘现象,而运动皮质神经元细胞完好,表明最初的改变产生于神经轴突的远端,逐渐向上逆行累及大脑中央前回的锥体细胞,此种改变又称为逆行性死亡。
一些生前仅有下运动神经元体征的ALS患者,死后尸检可见显著的皮质脊髓束脱髓鞘改变,表明前角细胞功能受累严重,掩盖了上运动神经元损害的体征。
还有一些临床表现典型的ALS,其病理改变类似于多系统变性,即有广泛的脊髓结构损害,脊髓前角、锥体束、脊髓小脑后束、脊髓后索的神经根间区、Clarke核以及下丘脑、小脑齿状核和红核均有神经元细胞脱失和胶质细胞增生。
采用免疫组织化学染色方法可以在中枢神经系统的不同部位的神经细胞发现异常的泛素阳性包涵体。
这些包涵体包括以下几种类型:
(1)线团样包涵体,电镜下包涵体为条索或管状,通常带有中央亮区为嗜酸或两染性。
被一淡染晕区包绕,在HE染色中不易见到。
(2)透明包涵体,为一种颗粒细丝包涵体。
细丝直径为15~20nm,颗粒物质混于细丝间形成小绒球样致密结构,外周常有溶酶体样小体及脂褐素等膜性结构包绕。
(3)路易体样包涵体,为一圆形包涵体,由不规则线样结构与核糖体样颗粒组成,中心为无定形物质或颗粒样电子致密物,这些物质包埋于18nm细丝中,排列紧密或松散,外周有浓染的环,类似路易体。
(4)Bunina小体,是ALS较具有特异性的病理改变。
这些包涵体主要分布于脊髓的前角细胞和脑干运动核神经细胞,也可以出现在部分运动神经元病患者的海马颗粒细胞和锥体细胞、齿状回、嗅皮质、杏仁核、Onuf核、额颞叶表层小神经元和大锥体细胞胞质中。
中医病因病机分析
该病起病隐袭,常无外感温热之邪,灼肺伤律的过程,大多一旦出现症状,便主要表现为虚损之象。
因此本病主要是由先天禀赋不足,后天失养,如劳倦过度,饮食不节,久病失
治等因素损伤脾胃肝肾,致气血生化乏源或精血亏耗,则筋脉肌肉失之儒养,肌萎肉削,发为本病。
(一)脾胃虚损
脾为后天之本,津液气血生化之源,主四肢肌肉主运化主涎;胃主受纳,饮食入胃。
游溢精气,上输于脾,脾气散精,上归于肺,布散于全身。
脾胃虚弱,或因病致虚,由虚致损、损伤脾胃,使脾胃受纳运化失常,气血生化不足,无以生肌,四肢不得禀水谷之气,无以为用,故出现四肢肌肉萎缩,肌肉无力,甚至吞咽困难,咀嚼无力,口张流涎。
脾虚累及肺脏、肺主气主声,故出现语音含糊,构音不清,呼吸气短。
(二)脾肾虚损
肾为先天之本,主藏精,精生髓,先天禀赋不足,精亏髓少,或劳倦伤肾,肾气亏虚,不能温煦脾阳,脾阳不振,不能运化水谷精微以濡养肌肉筋脉,即出现四肢肌肉萎缩、肢体无力。
肾为作强之官,肾气之充沛,又需脾胃之补养,脾肾两虚则骨枯髓虚,形瘦肉萎,腰脊四肢痿软无力。
(三)肝肾阴虚
肝藏血,主筋,主风,主动;肾藏精,主骨主髓。
先天不足,肾精亏虚,或房事不节,或劳役过度,精损难复,阴精亏损,虚阳浮动,肝血不能濡养筋脉,虚风内动,故见肌束颤动,肢体痉挛。
但凡肌肉震颤跳动,腰反射亢进者,责之于肝。
(四)湿热浸淫,虚实夹杂
脾土恶湿喜燥,肝脏体阴用阳,肺朝百脉,通调水道。
脾虚失运则聚湿化热,火与元气不两立,一胜则一负,由是阴火内炽。
阴火主要是下焦肝肾之火,肝经湿热浸淫,流注于下,筋骨萎软无力。
另外,脾胃虚弱,内生湿热,阻碍运化,精微物质不能上输于肺,百脉空虚,肌肉组织失养。
故本型为虚实夹杂之证。