核苷酸序列分析
- 格式:ppt
- 大小:3.18 MB
- 文档页数:32

寡核苷酸序列分析
寡核苷酸序列分析是一种用于研究和确定短的DNA或RNA片段(通常为20-30个核苷酸)核苷酸种类和排列顺序的生物技术,在许多生物学和医学领域都有重要的应用价值。
寡核苷酸是药物研究中的一个新热点,一些具有特异性结合或者裂解致病基因的寡核苷酸可以开发成新型药物,它们作用效率高、应用范围广,可以对传统药物进行补充,具有广泛的应用前景。
对寡聚核苷酸进行序列分析是基因药物的研发和生产过程中必不可少的一环。
BTP-寡核苷酸序列分析流程。
质谱技术的发展为寡核苷酸序列鉴定提供了强有力的分析方法。
寡核苷酸分子在高分辨率质谱仪中被解离成一系列碎片离子,这些碎片带有质荷比(m/z)值,通过对这些片段离子的质谱分析,可以推断原始寡核苷酸的碱基序列。
百泰派克生物科技(BTP)通过CNAS/ISO9001双重质量体系认证,公司基于Thermo公司的Obitrap Fusion Lumos质谱仪结合Nano-LC纳升色谱技术,开发并验证了高精度寡核苷酸序列分析方法,能够对ASO(反义寡核苷酸)、siRNA(小干扰RNA)、miRNA(微小RNA)、Aptamer(核酸适配体)等不同的寡核苷酸样品进行高分辨、高准确度的序列鉴定,满足客户不同的需求。
中/英文项目报告
在技术报告中,百泰派克会为您提供详细的中英文双语版技术报告,报告包括:
1. 实验步骤(中英文)。
2. 相关的质谱参数(中英文)。
3. 寡核苷酸序列分析详细信息。
4. 质谱图片。
5. 原始数据。
寡核苷酸序列分析一站式服务
您只需下单-寄送样品。
百泰派克一站式服务完成:样品处理-上机分析-数据分析-项目报告。

核苷酸序列比对与基因家族演化分析概述核苷酸序列比对和基因家族演化分析是生物信息学中重要的研究方法。
核苷酸序列比对是将两个或多个核苷酸序列进行比较,并通过寻找相似性和变异性来研究它们之间的关系。
基因家族演化分析则是通过比对相关基因的核苷酸序列,探究它们的进化历程和亲缘关系。
本文将详细介绍核苷酸序列比对和基因家族演化分析的原理、方法和应用。
核苷酸序列比对的原理与方法核苷酸序列比对是通过比较两个或多个核苷酸序列的完全性、相似性和变异性来推断它们之间的关系。
核苷酸序列比对的原理基于生物进化的基本思想:相同的DNA序列在不同物种中表现出不同的特征,这些特征可以反映物种之间的进化关系。
核苷酸序列比对的方法主要分为全局比对和局部比对两种。
全局比对适合于相似性较高的序列,它通过考虑整个序列的相似性来确定最佳比对位置。
局部比对则用于相似性较低的序列,它只关注具有较高相似性的区域,从而可以发现更多的共同特征。
核苷酸序列比对的常用算法包括Smith-Waterman算法和Needleman-Wunsch算法。
Smith-Waterman算法是一种局部比对算法,通过计算一个得分矩阵来找到最佳的匹配位置。
Needleman-Wunsch算法则是一种全局比对算法,它将序列比对问题转化为一个路径搜索问题,通过动态规划的方法找到最优路径。
核苷酸序列比对的应用非常广泛。
它可以用于研究同一物种内的个体间差异,如单核苷酸多态性(SNP)的分析。
此外,它还可以用于研究不同物种之间的亲缘关系,如物种分化和进化的研究。
基因家族演化分析的原理与方法基因家族演化分析是通过比对一组相关基因的核苷酸序列,研究它们的进化历程和亲缘关系。
基因家族是指具有共同起源的一组基因,它们在物种中以多个拷贝的形式存在。
基因家族演化分析的方法主要包括系统进化树构建和序列聚类分析。
系统进化树构建是通过比对一组相关基因的核苷酸序列,计算它们之间的距离或相似性,并将它们构建成一个进化树来描述它们的亲缘关系。


专利核苷酸序列表问题核苷酸序列是基因组学研究中的重要组成部分,它是描述DNA或RNA中碱基序列的方法。
通过分析核苷酸序列,可以研究和了解生物的遗传信息、物种间的相似性以及生物功能等。
在专利核苷酸序列表问题中,我们将探讨专利对核苷酸序列的保护以及可能涉及的一些问题。
在分析专利核苷酸序列问题之前,我们先来了解一下专利的基本概念。
专利是一种法律保护,给予发明者对其发明的独家权利,以鼓励创新和技术进步。
专利可以涵盖多种技术领域,包括生物技术、医药、化学等。
核苷酸序列专利是一种在生物技术领域中常见的专利类型。
核苷酸序列专利包括对DNA或RNA中的核苷酸序列进行保护。
在申请核苷酸序列专利时,需要提供足够的信息来证明该序列的独创性和实用性。
通常情况下,申请人需要提交核苷酸序列的具体序列以及相关的实验数据,以支持该序列在特定领域的应用。
此外,核苷酸序列专利还需要符合专利法的要求,如新颖性、创造性和工业适用性。
在核苷酸序列领域,新颖性通常是一个挑战,因为生物体中的许多核苷酸序列已经被发现并研究过。
因此,申请人需要证明其序列的新颖性。
创造性要求申请人应该展示其序列在特定领域的创新,即该序列具有明显的技术进步。
工业适用性要求申请人应该表明其序列在实际应用中具有一定的用途。
在申请核苷酸序列专利时,还需要注意保护范围的选择。
保护范围是指专利权的边界,即专利权益的范围。
在核苷酸序列专利中,保护范围可以是具体的核苷酸序列或该序列的变种。
申请人可以选择保护整个序列,也可以选择保护该序列的一部分。
不过,保护范围的选择需要谨慎考虑,以确保能够充分保护申请人的权益。
此外,核苷酸序列专利还涉及到数据公开和保密的问题。
核苷酸序列的专利申请通常需要公开,以便其他人能够了解和使用该技术。
然而,核苷酸序列的公开也存在一些问题。
首先,公开的核苷酸序列可能被他人使用,并申请专利。
其次,公开的核苷酸序列可能丧失商业机密性,从而可能导致竞争对手的技术窃取。

生物信息学中的DNA和RNA序列分析方法随着生物研究的发展,生物信息学逐渐成为了一个十分重要的学科领域,DNA和RNA序列分析是其中较为重要的一个方面。
DNA和RNA是生物体中的核酸,它们携带了生命的遗传信息,而对这些信息进行解读和分析就需要运用到生物信息学。
本文将为大家介绍生物信息学中的DNA和RNA序列分析方法。
一、基础知识在深入了解DNA和RNA序列分析方法之前,我们需要先了解一些基础知识。
1. DNA和RNA的基本结构DNA双链螺旋结构由核苷酸组成,其中核苷酸由磷酸、五碳糖核糖或脱氧核糖和一种氮碱基组成。
常见的氮碱基有腺嘌呤(A)、鸟嘌呤(G)、胸腺嘧啶(T)和胞嘧啶(C)。
RNA是由核苷酸组成的单链分子,比DNA少了胸腺嘧啶,而是由尿嘧啶(U)取代了。
2. DNA和RNA的编码DNA编码了基因信息,而RNA通过转录形成mRNA,再到翻译形成蛋白质。
在转录过程中,mRNA中的氮碱基按照特定的规则与DNA上的氮碱基匹配,即腺嘌呤与尿嘧啶配对,鸟嘌呤与胞嘧啶配对。
这种配对方式被称之为互补配对。
RNA与DNA的互补配对非常重要,因为它决定了RNA能够识别和复制DNA中的信息。
二、DNA和RNA序列分析方法DNA和RNA序列分析方法主要有以下几种。
1. 序列比对序列比对是指将两个或多个序列进行比较,找出它们之间的相似处和差异。
序列比对是进行生物信息学研究的基础,也是DNA 和RNA序列分析的核心方法。
序列比对有两种类型,全局比对和局部比对。
全局比对一般用来比较两个完整的序列,例如蛋白质序列。
局部比对一般用来比较一个序列中的一小段与另一个序列中的一小段。
2. 序列注释序列注释是指将序列上的功能信息注释到序列上。
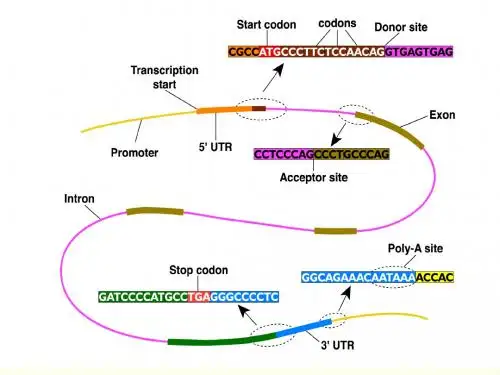
一般情况下,序列注释会包括以下几个方面的信息:基因结构,包括外显子、内含子、UTR等;转录因子结合位点、启动子和增强子等调控元件;蛋白质结构,包括功能和结构域等;翻译起始和终止位点等。
序列注释需要利用已知的信息,例如已知的基因、蛋白质和调控元件等数据库信息。



核酸序列分析1、核酸序列检索可通过NCBI使用Entrez系统进行检索,也可用EBI的SRS服务器进行检索。
在同时检索多条序列时,可通过罗逻辑关系式按照GenBank接受号进行批量检索。
如用“AF113671 [ac] OR AF113672 [ac]”可同时检索这两条序列。
其中“[ac]”是序列接受号的描述字段。
2、核酸序列的基本分析(1)分子质量、碱基组成、碱基分布分子质量、碱基组成、碱基分布可通过一些常用软件等直接获得。
如:BioEdit(/BioEdit/bioedit.html),DNAMAN()。
(2)序列变换进行序列分析时,经常需要对DNA序列进行各种变换,例如反向序列、互补序列、互补反向序列、显示DNA双链、转换为RNA序列等。
这些用DNAMAN软件可很容易实现,这些功能集中在Sequence→Display,从中可选择不同的序列变换方式对当前通道的序列进行转换。
(3)限制性酶切分析该方面最好的资源是限制酶数据库(Restriction Enzyme Database,REBASE)。
REBASE数据库(,/rebase)中含有限制酶的所有信息,包括甲基化酶、相应的微生物来源、识别序列位点、裂解位点、甲基化特异性、酶的商业来源及公开发表的和未发表的参考文献。
其它资源还有:WebGene:/~tjyin/WebGene/RE.html,/personal/tyin.htmlWebCutter2:http://www//firstmarkert/firstmarket/cutter/cut2.html同时,很多软件也能够识别REBASE限制酶数据库。
强烈推荐使用集成化的软件如BioEdit和DNAMAN等。
所得出的结果给出指定DNA序列的酶切位点信息,为克隆鉴定和亚克隆提供了重要信息。
在实际进行分子生物学实验中,有时需要对多条相关序列(如发生突变的一批序列)同时进行酶切分析,以便为后续的克隆鉴定提供参考。

核苷酸序列比对与进化分析核苷酸序列比对与进化分析是一种用于研究生物界的进化历程和分子关系的重要方法。
通过比对不同生物个体之间的核苷酸序列,我们可以揭示它们在进化过程中的关系,并推测它们之间的共同祖先。
本文将介绍核苷酸序列比对的原理和常用工具,并探讨如何进行进化分析。
这项技术在生物学研究中具有重要的应用前景。
核苷酸(DNA或RNA)是生物体内以脱氧核苷酸或核苷酸为基本单位组成的分子。
每个个体的DNA或RNA序列都是独特的,并且在进化过程中会发生突变和改变。
通过比对不同个体之间的核苷酸序列,我们可以发现它们之间存在的相似性和差异性。
相似的核苷酸序列可能表示着共同的祖先,而差异的核苷酸序列则可能反映了它们之间的进化过程。
核苷酸序列比对是通过比较两个或多个核苷酸序列以确定它们之间的相似性的过程。
它通常分为全局比对和局部比对两种类型。
全局比对适用于相似序列的比对,而局部比对则适用于不完全相似或相关的序列的比对。
核苷酸序列比对的目的是找出序列之间的同源性区域,也就是彼此相似的片段。
在比对过程中,我们可以使用不同的算法和工具,如BLAST、ClustalW和MAFFT等,来进行序列比对。
BLAST(Basic Local Alignment Search Tool)是一种常用的用于核苷酸序列比对的工具。
它可以在数据库中搜索与查询序列相似的序列,并计算它们之间的相似度得分。
BLAST使用两个关键指标来评估序列的相似性:E值和比对得分。
E值表示在以随机事件为基础的模型下,获得当前比对得分的预期频率。
较低的E值表示较高的相似性。
比对得分则用于表示比对序列的相似性程度,通常以位分数(bitscore)的形式呈现。
ClustalW是一种常见的多序列比对工具,它可以将多个序列比对为一个较长的序列。
ClustalW将序列的相关部分放在一起,从而构建一个类似于进化树的序列比对结果。
同时,ClustalW还通过计算保守性指数,识别出进化或功能上重要的区域。

全基因组关联研究名词解释
全基因组关联研究(Genome-wide Association Study,GWAS)是一种用于识别与特定疾病或表型相关的基因变异的强大工具。
下面是对全基因组关联研究涉及的一些关键术语的解释:
1. 基因组作图(Genome Mapping):
基因组作图是指构建DNA序列或基因位置的精细图谱的过程。
这涉及到识别和定位基因、DNA序列及染色体上的各种特征。
基因组作图是全基因组关联研究的基础,因为它可以帮助科学家确定与特定疾病或表型相关的基因变异的确切位置。
2. 核苷酸序列分析(Nucleotide Sequencing):
核苷酸序列分析是确定DNA序列的过程,它有助于识别基因变异和等位基因。
通过核苷酸序列分析,科学家可以识别出与特定疾病或表型相关的等位基因,并理解它们如何影响生物体的性状和功能。
3. 基因定位(Gene Mapping):
基因定位是指确定基因在染色体上的位置的过程。
通过基因定位,科学家可以确定与特定疾病或表型相关的基因的确切位置。
这有助于揭示基因变异如何影响疾病的发生和发展。
4. 时序表达模式和基因功能分析(Temporal Expression Patterns and Gene Function Analysis):
时序表达模式分析涉及确定基因在生物体生命周期的不同阶段中的表达情况。
这有助于理解基因在特定疾病或表型中的作用,并揭示其在生物过程中的功能。
通过基因功能分析,科学家可以了解特定基因
变异如何影响生物体的生理和生化过程。
这些分析有助于深入了解疾病的发病机制和潜在的治疗方法。
氨基酸序列和核苷酸序列的关系氨基酸序列和核苷酸序列是生物学中常用的两种序列。
氨基酸序列指的是多肽链中氨基酸的排列顺序,而核苷酸序列是指DNA或RNA中核苷酸的排列顺序。
这两种序列在生物学研究中具有重要的意义,可以通过比对和分析序列来揭示生物体的结构和功能。
氨基酸序列是蛋白质的基本组成单位。
蛋白质是生物体内功能最为复杂和多样的分子,它们参与了几乎所有生物过程。
蛋白质的功能主要由其氨基酸序列决定,不同的氨基酸序列可以使蛋白质具有不同的结构和功能。
通过对氨基酸序列的研究,可以揭示蛋白质的结构和功能,以及蛋白质与疾病之间的关系。
核苷酸序列是DNA和RNA的基本组成单位。
DNA是生物体遗传信息的储存介质,而RNA则在遗传信息的转录和翻译过程中起到重要的作用。
核苷酸序列的分析可以揭示DNA和RNA的结构和功能,以及遗传信息的传递和表达。
通过对核苷酸序列的比对和分析,可以推断基因的功能和进化关系,同时也可以研究疾病与基因之间的关系。
氨基酸序列和核苷酸序列之间存在着密切的关系。
在生物体内,氨基酸序列是由核苷酸序列编码的。
DNA中的每三个核苷酸对应一个氨基酸,这被称为密码子。
不同的密码子对应不同的氨基酸,这样就可以通过核苷酸序列推导出氨基酸序列。
这一过程称为转录和翻译,是生物体遗传信息的表达和实现过程。
在转录过程中,DNA的双链解旋,mRNA链与DNA链互补配对,形成mRNA的单链。
mRNA链上的核苷酸序列与DNA链上的核苷酸序列一一对应,但在mRNA中,腺嘌呤(A)被尿嘧啶(U)取代。
这样,DNA中的T(胸腺嘧啶)与mRNA中的A(腺嘌呤)对应,A与U互补配对。
转录过程中,mRNA的核苷酸序列与DNA的核苷酸序列是一一对应的。
在翻译过程中,mRNA链被核糖体扫描,通过tRNA带有的氨基酸与mRNA上的密码子互补配对,从而将氨基酸连成多肽链。
tRNA 中的核苷酸序列与mRNA中的密码子核苷酸序列互补配对,从而将氨基酸按照正确的顺序连接起来。
核苷酸序列分析软件工具核苷酸序列分析是生物学研究中的重要环节,它可以帮助科学家们更好地理解生物体内的基因组成,以及基因在生物体内的功能。
为了使这一分析过程更加高效和准确,科学家们开发了一系列核苷酸序列分析软件工具。
本文将介绍几个常用的核苷酸序列分析软件工具,并对它们的特点和功能进行详细的解析。
首先,我们来介绍一款被广泛使用的核苷酸序列分析软件工具,即NCBI BLAST(Basic Local Alignment Search Tool)。
NCBI BLAST是一款免费的基因序列比对工具,它能够比对用户提供的核苷酸序列与已知数据库中的序列进行比对,并给出最相似的序列。
通过NCBI BLAST,科学家们可以快速地找到与自己研究对象相关的基因序列,并对它们进行进一步的分析。
NCBI BLAST具有高效、准确的特点,广泛应用于基因组学、生物信息学等领域。
另一个常用的核苷酸序列分析软件工具是DNASTAR Lasergene。
DNASTAR Lasergene是一套全面的分子生物学和生物信息学软件套件,它包含了多个功能强大的软件模块,可以进行多样化的核苷酸序列分析工作。
其中,SeqBuilder模块可以帮助用户进行序列的编辑和构建;序列比对模块可以进行多序列比对和进化分析;序列注释模块可以对序列进行注释和功能预测等。
DNASTAR Lasergene具有界面友好、操作方便等特点,适用于不同层次的用户。
此外,我们还有Geneious作为一个强大的核苷酸序列分析软件工具。
Geneious提供了全面的基因组和蛋白质序列分析功能,可以进行序列比对、进化分析、结构预测、序列注释等多种操作。
与其他软件相比,Geneious具有更强的计算能力和更丰富的功能。
它也支持自定义插件的添加,用户可以根据自己的需求扩展软件的功能。
Geneious在生物学研究领域中被广泛使用,尤其在基因组和序列分析的研究中具有很高的声誉。
此外,还有一些核苷酸序列分析软件工具适用于特定的研究领域。
dna鉴定方法DNA鉴定(DNA Identification)是一种比对技术,用于将两份或两份以上的DNA样本进行特定分子位点(Marker)的比对,以确认其是否来自同一个人,或者从同一个祖先血统。
目前DNA鉴定方法主要有多态性位点分型方法(Polymorphic Marker)、核苷酸序列分析方法(Nucleotide Sequencing)、基因组测序(Genome Sequencing)和周期温和随机引物扩增方法(PGR-PCR)等。
①多态性位点分型方法:多态性位点分型方法是一种利用DNA变异性手段在实验中进行比对的方法,主要通过在DNA序列上的泛稀疏多态性,将适当的位点作为鉴定依据,从而实现独特的基因型识别,将两个或更多个DNA样本进行比对,以鉴定出它们之间的相似性或不同性。
其中,最常用的技术有STR技术(short tandem repeat)、VNTR技术(variable number of tandem repeat)和AFLP技术(amplified fragments length polymorphism)。
②核苷酸序列分析方法:核苷酸序列分析方法是采用生物信息学手段,对DNA片段的核苷酸序列进行比对和分析,从而鉴定出相似或不同之处,判断两组样本是否具有相同的祖先血统,从而进行身份鉴定。
该方法可以在较短时间、较少费用内得到准确的结果,是一种高效可靠的身份鉴定技术方法,主要技术有基因测序(Gene Sequencing)、单碱基多态性分析(Single Nucleotide Polymorphism Analysis)和全基因组测序(Whole Genome Sequencing)。
③基因组测序:基因组测序也称作全基因组测序,是指对物种的整个基因组进行双向测序,以确定基因组序列及其组成结构的技术。
它是基于大规模的DNA测序,可以比较鉴定出样本间的基因序列差异,从而回答遗传学与种群学研究问题。
基因的序列分析基因是生命体中的基本单位,控制着生物体的发育、生长和繁殖等过程。
通过对基因序列的分析,可以有效了解这些基本单位的功能和变化,从而为生命科学的研究和相关应用提供基础支持。
本文主要介绍基因的序列分析,包括基本概念、主要方法和相关应用等方面,以期为读者提供一些参考和启示。
基因序列的基本概念基因序列是指一条由核苷酸(DNA或RNA)组成的线性序列,是表达基因信息的物质基础。
天然基因序列通常以ATCG(DNA)或AUCG(RNA)四种字母作为基本单元,组成一些特定的字符串,例如“ATGACAAGCTTCTCAGTCAAGG”就代表了一个简单的DNA序列。
基因序列的长度可以非常巨大,微生物基因有数百个核苷酸,而人类基因的长度则通常在数万个核苷酸到数百万个核苷酸之间。
基因序列可以分为编码区和非编码区,其中编码区包含了编码蛋白质的基因的信息,而非编码区则包含了调节元件、基因启动子、转录因子结合位点等信息。
基因序列的分析方法直观分析法最原始、最简单的基因序列分析方法,是通过人工直接查看基因序列,了解其中蕴含的信息。
这种方法最常用于微生物遗传学研究中,早期的遗传学家利用这种方法,解析了许多微生物路径方式和代谢途径的信息。
但是这种方法存在着许多缺陷,例如需要繁琐耗时地逐个查看碱基,对于长度较长的基因序列来说,不仅容易犯错,而且很难发现潜在的模式和规律。
计算机分析法随着计算机科学的发展,基因序列的计算机分析方法也得到了广泛应用。
为了更好地描述基因序列,科研工作者将碱基序列转换为字符串,并进行序列分析和比对。
目前,计算机分析方法主要包括序列比对、序列聚类、序列模式识别等几个方面,具体如下:1.序列比对分析序列比对分析是将不同物种的基因序列进行比对,找出两方之间的相似点和差异点。
一方面可以为进化分析和生物系统学研究提供基础支持,另一方面还可以通过比对得到基因的同源模板序列和保守区域序列等信息。
2.序列聚类分析序列聚类分析是将基因序列进行分类,并划分出相互关系紧密、同源性大的序列群。