地高辛标记探针在Southern杂交分析中的技术要点
- 格式:pdf
- 大小:210.17 KB
- 文档页数:2

地高辛标记探针在Southern杂交分析中的技术要点地高辛标记探针在Sou thern杂交分析中的技术要点陆小平周文军(苏州大学生命科学学院江苏苏州215006)小岛峰雄(日本信州大学纤维学部)外源基因是否成功导入受体材料的基因组中,必须从转化植株中找到分子生物学的检测证据。
目前,PCR、Southern杂交等作为常用检测手段而被广泛采用。
虽然PCR技术可以快速得到结果,但是,以农杆菌介导的材料必须慎重这一结论,以免由于农杆菌污染造成假阳性。
而Southern分析由于操作程序繁琐,对植物基因组DNA的提取、纯化、酶切等技术要求较高,有时使杂交结果不甚理想。
我们在植物基因转化的研究中,对荞麦,桑树,紫景天,洋麻等基因组DNA的提取、纯化、酶切进行了探讨,对流程中的有关步骤进行技术改良,使酶切后的PNA在凝胶板上是涂布状完全达到了Southern杂交的技术要求,并用地高辛(D ig)标记探针对外源DNA进行分析,取到了较好的效果。
现简述如下∶1 植物基因组D NA的提取及纯化1)提取高纯度的DNA是Southern杂交的关键, DNA的粗提物中,往往含有大量的蛋白质、多糖、单宁、色素等大分子杂质。
这些杂质通常与DNA共同沉淀或与DNA聚合成大分子复合物。
一旦复合物形成,即便在以后的操作中用酚、氯仿多次纯化,也很难将其除去。
我们的经验是:当用液氮破碎新鲜样品的细胞壁后,先用蒸馏水洗脱两次(洗脱温度为42℃),每次5m in。
以达到洗脱多糖的目的。
每次洗脱后离心5m in(13000 r m in),弃去上层液。
该上层液中含有粘度较高的胶状体,其主要成分可能是粘性多糖。
在提取桑树基因组DNA时,最好用叶柄或幼茎、幼叶作提取材料。
在样品有限时,也可用成熟叶甚至老叶代替。
据C lark M S (1998)介绍,取样前对材料进行除淀粉处理(减少光照、黑暗处理24h),可以抑制多糖的污染。
但我们采用除淀粉处理后的实验结果并不理想,其效果远不及用蒸馏水洗脱。
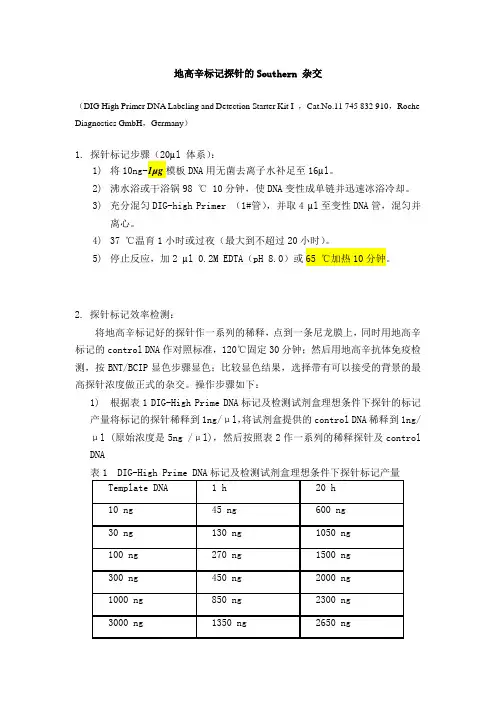
地高辛标记探针的Southern 杂交(DIG High Primer DNA Labeling and Detection Starter Kit I ,Cat.No.11 745 832 910,Roche Diagnostics GmbH,Germany)1. 探针标记步骤(20µl 体系):1)将10ng-1µg模板DNA用无菌去离子水补足至16µl。
2)沸水浴或干浴锅98 ℃ 10分钟,使DNA变性成单链并迅速冰浴冷却。
3)充分混匀DIG-high Primer (1#管),并取4 µl至变性DNA管,混匀并离心。
4)37 ℃温育1小时或过夜(最大到不超过20小时)。
5)停止反应,加2 µl 0.2M EDTA(pH 8.0)或65 ℃加热10分钟。
2. 探针标记效率检测:将地高辛标记好的探针作一系列的稀释,点到一条尼龙膜上,同时用地高辛标记的control DNA作对照标准,120℃固定30分钟;然后用地高辛抗体免疫检测,按BNT/BCIP显色步骤显色;比较显色结果,选择带有可以接受的背景的最高探针浓度做正式的杂交。
操作步骤如下:1)根据表1 DIG-High Prime DNA标记及检测试剂盒理想条件下探针的标记产量将标记的探针稀释到1ng/μl,将试剂盒提供的control DNA稀释到1ng/μl (原始浓度是5ng /μl),然后按照表2作一系列的稀释探针及control DNA表1 DIG-High Prime DNA标记及检测试剂盒理想条件下探针标记产量表2)将上述稀释的2-9 号管的control DNA 及探针DNA各取1μl点膜3)120℃固定30分钟或紫外交链3-5分钟4)将膜放入装有20ml Maleic acid buffer 的塑料器皿中,室温振荡2分钟5)将膜放入10ml Blocking solution中室温温育30分钟6)将膜放入10ml Antibody solution 中室温温育30分钟7)用10ml Washing buffer 洗2次,每次15分钟8)在10ml Detection buffer 平衡2-5分钟9)将膜放入2ml 新配制的Color substrate solution 中暗室条件下显色。

Southern Blot原理及实验方法Southern Blot原理及实验方法原理:将待检测的DNA分子用/不用限制性内切酶消化后,通过琼脂糖凝胶电泳进行分离,继而将其变性并按其在凝胶中的位置转移到硝酸纤维素薄膜或尼龙膜上,固定后再与同位素或其它标记物标记的DNA或RNA探针进行反应。
如果待检物中含有与探针互补的序列,则二者通过碱基互补的原理进行结合,游离探针洗涤后用自显影或其它合适的技术进行检测,从而显示出待检的片段及其相对大小。
用途:检测样品中的DNA及其含量,了解基因的状态, 如是否有点突变、扩增重排等。
试剂和器材一、试剂变性液:1.5mol/L NaCl,0.5mol/L NaOH。
中和液:0.5mol/L Tris-HCl (pH=7.5),1.5mol/L NaCl。
20×SSC:3mol/L NaCl,0.3mol/L 柠檬酸钠。
以上溶液均在100Kpa灭菌20分钟。
2×SSC:用无菌移液管吸取20×SSC溶液5mL,加无菌水45mL。
6×SSC:用无菌移液管吸取20×SSC溶液15mL,加无菌水75mL。
二、器材22cm×15cm瓷盘操作方法1. 在琼脂糖凝胶上电泳分离DNA。
取出凝胶,切去边缘多于部分,EB染色,在紫外灯下照相(放一标尺,可从像片中读出DNA迁移的距离)。
2. 将凝胶置于200mL变性液中,浸泡45分钟,并温和地不断振荡,使凝胶上的ds-DNA转变为ss-DNA,然后用重蒸水冲洗凝胶几次。
3. 用中和液浸泡凝胶并不断地振荡45分钟,将凝胶中和至中性。
防止凝胶的碱性破坏硝酸纤维膜。
4. 取一个瓷盘,在底部放一块玻璃板(或一块海绵)使盛器内的20倍SSC转移滤液低于玻板表面,在玻板表面盖一张3mm的二号滤纸,滤纸两边浸没于20倍SSC溶液中,在玻璃和滤纸之间,赶掉所有的气泡。
5. 把凝胶底面朝上放在滤纸上,赶走两层之间出现的气泡。

Southern 印迹杂交实验报告姓名:陆叶学号:14211020062 专业:公共卫生实验时间:12.18;12.25 带教老师:潘銮凤【实验目的】学习核酸杂交的基本过程和操作【实验原理】将待检测的DNA分子用或不用限制性内切酶消化后,通过琼脂糖凝胶电泳进行分离,继而将其变性并按其在凝胶中的位置转移到硝酸纤维素薄膜或尼龙膜上,固定后再与同位素或其它标记物标记的DNA或RNA探针进行反应,检测靶DNA片段中是否存在与探针同源的序列。
如果待检物中含有与探针互补的序列,则二者通过碱基互补的原理进行结合,游离探针洗涤后用自显影或其它合适的技术进行检测,从而显示出待检的片段及其相对大小。
用途:检测样品中的DNA及其含量,了解基因的状态, 如是否有点突变、扩增重排等。
【实验步骤】1、基因组DNA的限制性内切酶酶切取1个1.5ml离心管按下列量依次加入基因组DNA、10X酶缓冲液,酶,做好标记。
老师给的HL60基因组DNA 10μg(7.6μL);10×Buffer E(10.4μL);EcoR I(10 u/μL)(2μL)。
总体积20μL。
37℃保温1.5小时。
2、1%琼脂糖电泳先制备凝胶,称取1.2 g Agarose放入三角烧瓶中,加入60 mL TAE溶液,微波炉中加热令其完全溶解,稍作冷却后加入GelRed核酸染液6μL(稀释10,000倍)。
浇板,水平放置,待凝。
胶凝后按照以下顺序加样。
两组共用一块凝胶,共7个样。
①基因组DNA10 20μl (自己的)②基因组DNA10 μg(老师的)③酶切样品④阳性对照(c-myc 4.7 kb线性片段,30pg/μl, 10μl)⑤酶切样品⑥基因组DNA10 μg (老师的)⑦基因组DNA10 20μl (自己的)插上电源,负极在样品槽一边,DNA将向阳极跑,80V电泳2小时左右,直到溴酚蓝染料跑到接近凝胶尾部,在电泳期间做好胶变性和转移准备。

Southern 杂交试验方案一、探针标记1、探针标记严格按照罗氏试剂盒说明书进行;2、标记探针检测:取标记产物和普通PCR扩增产物各1μl,1.0%的琼脂糖凝胶电泳。
由于大分子量的DIG掺入,标记产物泳动速度比普通PCR产物要慢。
所以根据电泳图就可以判断是否标记上和有没有非特异标记!其余标记产物-20℃保存。
如果要准确检测标记效率(一般不必),取标记产物1μl和Dig标记的对照DNA,用DNA 稀释液按10倍系列稀释后点样于带正电荷尼龙膜上。
用紫外交联仪或真空烘烤固定DNA探针,按杂交检测方法进行信号检测,然后与膜条上的Dig标记的对照DNA信号强度对比,推算出标记的探针浓度。
如初始模板量较大,可取1μl标记产物稀释10~100倍后定量。
二、基因组DNA酶切1、采用EcoRI和一对甲基化敏感同裂酶HPaIl、MspI消化高粱基因组DNA 5ug,3 Unit/ug DNA,50uL反应体系,在37℃条件下酶切消化10h。
2、酶切产物经1%琼脂糖凝胶电泳(电压小于1v/cm,12h)分离。
凝胶在0.2mol/L的HCl 中脱嘌呤处理,至溴酚蓝变为黄色,弃去HCl后用蒸馏水洗几次,然后以0.4mol/L的NaOH 为转移液,利用毛细管作用将DNA转移到Hybond N+尼龙膜上,转移过夜后将膜取出夹放于滤纸间,80℃烘烤2小时。
常温放于干燥处保存备用。
(转膜:1、将胶裁成9.8cm×8.0cm 大小,于平皿中用蒸馏水冲洗一次;(此步一定记好胶的大小,后面裁纸/膜以及杂交液体积的选择都要用到)2、加入100ml 0.25 mol/L 的盐酸脱嘌呤,室温振荡15~30 min,至溴酚蓝完全变成黄色。
(如果限制性片段>10kb,酸处理时间可适当延长;若限制性片段很小,此步可省略)3、用蒸馏水冲洗2 次。
加入变性液(1.5mol/L NaCl、0.5mol/L NaOH)室温振荡20~30min;至溴酚蓝完全恢复到原来的蓝色。


地高辛标记探针的DNA印迹方案Southern blot using DIG Prober(分子生物学实验室)Step I 采用地高辛高效标记混合物(ROCHE) 标记DNA探针1 以质粒为模板,PCR扩增序列特异性片段,回收片段,并测定浓度浓度测定:将回收产物取1μl稀释5倍,标准分子量的标记物MAKER分别点1μl,2μl,4μl,6μl.然后比较回收产物的条带亮度与MAKER比较后,可以粗略估算浓度。
2 取1μg模板(第1步扩增回收产物)于1.5ml的eppendorf管中,加入灭菌双蒸水至终体积为16μl。
3 沸水浴处理10min后迅速于冰上冷却使DNA变性(1)摇匀DIG-High Prime(vial 1),取4μl加入已热变性的模板DNA中,混匀后,瞬时离心,37℃保温20h,65℃处理10min终止反应。
4取1μl电泳检测,标记后的条带稍大于标记前片断。
5标记好的DNA探针-20℃保存。
Step II 基因组DNA酶切1 提取高质量的基因组DNA,浓度>1μg/μl ; 测定浓度的方法同测定探针浓度方法一样。
2 选择合适的内切酶(酶切的DNA量20μg左右。
注意考虑到纯化的损失,约30%,酶切DNA浓度过大时,可以考虑多做几管做浓缩)。
50μl酶切反应体系:EcoR I (15U/μl ) 5μl10×M buffer 5μlgDNA 10μl(约10-20μg)ddH2O up to 50μl注:有些内切酶需加BSA,请参照内切酶说明(包括最适酶切温度)。
3 37℃酶切12h (电泳检测),视酶切情况可延长酶切时间,直至酶切完全。
65℃保温10min停止酶切反应。
Step III 预电泳和电泳1 配制0.8 %的琼脂糖凝胶(大胶40ml左右,胶薄较好,利于转膜)。
2 加入适量上样缓冲液(Loading Buffer)点样,点上质粒做对照。
注:两边的点样孔尽量空出来;点上marker,再单独一点样孔用溴酚兰做指示,防止跑过。

地高辛标记探针的Southern 杂交技术根据核酸变性与复性的特性,特定核苷酸序列的单链DNA或RNA能够在一定条件下与具有互补性的核酸链退火形成双链结构,称之为核酸杂交。
将已知的特定核苷酸序列的单链DNA或RNA进行标记,制成核酸探针,就可以用它检测样品中是否存在同源序列,或确定不同物种之间的亲缘关系,已成为分子生物学中一类重要的检测手段,在科学研究和实践中具有十分广泛的用途。
它可用于基因组特定DNA序列的定位,测定相关片段的同源性、从cDNA文库、基因组文库中筛选完整基因等。
还可以用于构建DNA分子的酶切图谱和遗传图—指纹分析等。
核酸杂交检测都是在滤膜上进行的,常用的滤膜有尼龙滤膜、硝酸纤维素滤膜等。
其基本过程包括以下几步。
首先将待检样品DNA或RNA分子直接点加到滤膜上,或经过凝胶电泳分离再通过毛细管作用或电导作用被转移到滤膜上,而且是按其在凝胶中的位置原封不动地“吸印〞上去的,这个过程称为核酸印迹〔nucleic acid blotting〕转移,主要有电泳凝胶核酸印迹法、斑点和狭线印迹法〔dot and slot blotting〕、菌落和嗜菌斑印迹法〔colony and plaque blotting〕;然后将具有核酸印迹的滤膜同带有放射性标记或其它标记的DNA或RNA探针进行杂交。
最后,经过放射自显影技术或光化学、免疫学技术显示出不同的颜色,根据颜色的有无和深浅判定结果。
根据操作方式和检测对象的不同,核酸杂交技术主要有以下几种:斑点杂交技术〔Dot blot〕、Southern杂交技术〔Southern blot〕、Northern杂交技术〔Northern blot〕。
前两者用于检测DNA,后者用于检测RNA。
本实验主要介绍Southern杂交技术。
1主要内容1. Southern Blot方法的原理。
2. DNA琼脂糖凝胶电泳。
3. DNA转移至硝酸纤维素膜/尼龙膜与膜处理。

分子生物学实验的常见问题与解决方案范文一、Southern杂交问题1:电泳后发现凝胶中DNA扩散,导致结果难以确定,如何解决这一问题?解决方案:(1)在操作上:电泳时隔孔上样,电泳后要对凝胶及时处理使凝胶干燥;(2)琼脂糖的质量应该较好,尤其是不应含有内切酶,否则有时将使低拷贝数基因的杂交结果难以解释。
问题2:传统方法中转膜不完全的问题如何克服?解决方案:经典的向上转移法会使凝胶短时间内变薄,此时即使延长转移时间至24小时以上,也不能使大分子DNA良好地转移出去,因此,转膜不完全。
向下转膜法由于不需在吸水纸上增加重量,凝胶基本不变形;加之吸水纸的吸力与水受到的重力方向一致,故可以良好地完成DNA的转移,它不需要特殊的仪器,转移速度较快,转移效率高。
利用地高辛标记探针进行Southern杂交时,对于大于15kb的DNA片断,转膜之前用盐酸进行脱嘌呤可以促进转膜效率,但脱嘌呤过度可导致分子量相对较小的DNA被打断成过小的片段而难以和尼龙膜结合。
如果实验转膜过程中同时含有大于15kb的DNA(通常为基因组)和小片段DNA(通常是基因组酶切产物),那么在转移的过程中倾斜容器,仅使凝胶上半部分浸泡于盐酸,这样既可以提高大片断DNA的转移效率,又不会打碎小片段DNA。
另外,等采用琼指糖凝胶直接杂交的方法,来解决这一难题:以转基因鼠的检测为例,实验步骤如下:1.转基因鼠的建立:以鼠乳清酸蛋白(WAP)基因5’调控区指导人G-CSF基因为构件,建立转基因小鼠。
转基因小鼠的检测采用剪取鼠尾DNA做Southern进行鉴定。
2.琼脂糖凝胶直接杂交:(l)用BanHI(150U)对由假孕鼠产生的仔鼠剪尾提取基因组DNA10μg酶切过液,取少量电泳检查酶切完全后,上样电泳8小时,(2)将电泳槽板连同凝胶置50℃放置3小时,用镊子轻轻揭起凝胶,放于变性液(0.5MNaOH,0.5MNaCl)20分钟,转置中和液(0.5MTriHCl,0.15MNaCl)20分钟,将胶放于玻璃板上沥干液体,室温放置30分钟。

分子杂交分子杂交是指在分子克隆中的一类核酸和蛋白质分析方法,用于检测混合样品中特定核酸分子或蛋白质分子是否存在,以及其分子量大小。
根据其检测对象的不同可分为 Southern 杂交、Northern 杂交和 Western 杂交,以及由此而简化的斑点杂交、狭线杂交和菌落杂交等。
一、 Southern blotting1.转膜当目标 DNA 经过限制性酶切并通过琼脂糖凝胶电泳以后,在 0.4N NaOH 碱性条件下变性,再在 1.5M NaCl/1M Tris(pH7.4)条件下中和使 DNA 仍保持单链状态。
然后通过毛细管渗吸或电转移或真空转移的方式,将凝胶上的 DNA 转移到硝酸纤维素滤膜或尼龙膜上。
最后通过80℃ 处理或紫外线照射将 DNA 固定在滤膜上。
图4-2是通过毛细管渗吸法进行 Southern blotting 的经典装置图。
2.分子杂交2.1 预杂交将结合了 DNA 分子的滤膜先与特定的预杂液进行预杂交,也就是将滤膜的空白处用鱼精 DNA 或牛奶蛋白封闭起来,防止在杂交过程中滤膜本身对探针的吸附。
2.2 杂交在一定的溶液条件和温度下,将标记的核酸探针与滤膜混合,如果滤膜上的DNA 分子存在与探针同源的序列,那么探针将与该分子形成杂合双链,从而吸附在滤膜上。
2.3 洗膜经过一定的洗涤程序将游离的探针分子除去。
3.检测3.1 放射性标记、检测在分子克隆操作中用于标记核酸分子的标记物主要是放射性同位素,如32P,33P和35S 。
在掺入核苷酸的标记过程中,只有三磷酸核苷酸的α位磷酸整合到核酸链中,因此使用α位磷酸被标记的三磷酸核苷酸,如 [ α- 32 P]dATP 。
而在标记 5' 末端磷酸基团的反应中一般使用γ位磷酸被标记的ATP 。
放射性的信号通过X- 光片放射自显影或磷屏扫描获取。
3.2 非放射性标记、检测。
其中应用最成功的是原德国宝灵曼公司开发的地高辛(digoxygenin, DIG)标记核酸探针。

生物技术通报BIOTECHNOLOGYBULLETIN・技术与方法・2008年第3期收稿日期:2007-11-19基金项目:国家自然科学基金(No.30460008)作者简介:刘立鸿(1982-),男,硕士生,生化与分子生物专业,E-mail:xjliulihong1234@126.com通讯作者:马正海(1971-),男,副教授,硕士生导师,E-mail:mzhxju@sohu.com自1975年英国科学家E.M.Southern创立Southern杂交技术以来,该技术已成为检测特定DNA片段的经典杂交方法之一[1]。
此法快速、准确、灵敏,目前已经广泛地应用于医学、病毒学、转基因动植物鉴定、动物疾病诊断以及DNA指纹分析等方面的研究。
Southern印迹杂交中使用的标记探针有同位素与非同位素标记2种。
放射性同位素标记的探针灵敏度较高,但存在半衰期限制,对操作者和环境会造成放射性辐射危害。
使用非同位素标记探针可避免放射性危害[2],常规实验室大多采用后者,其中最常用的是地高辛标记探针[3]。
目前虽然有地高辛标记探针标记的试剂盒,但是试剂盒侧重于步骤流程描述,关于技术要点的介绍不是很多。
国内关于地高辛标记探针Southern印迹杂交法技术要点报道也较少。
在开展Southern印迹杂交中,参照文献并结合实验室的具体情况对该技术的具体步骤进行了一些探索和改进,现作一总结。
1技术要点1.1地高辛探针的标记与使用1.1.1探针的标记方法标记方法有缺口平移法、末端标记法、随机引物法和聚合酶链反应法(PCR)等。
Southern杂交探针的制备一般用随机引物法,模板量应达到1μg以上,模板量不足1μg,可适当延长37℃温育时间。
当模板序列已克隆在载体上,地高辛标记探针Southern印迹杂交技术要点及改进刘立鸿1许璐1汪凯2张富春1马正海1(1新疆大学生命科学与技术学院分子生物学重点实验室新疆生物资源基因工程重点实验室,乌鲁木齐830046;2中国科学院上海巴斯德研究所,上海200025)摘要:Southern印迹杂交技术是检测特定DNA片段的常规方法之一,其操作程序繁琐,对基因组DNA的提取、纯化、酶切等均有较高要求,要获得理想杂交结果需要反复摸索。
地高辛标记核酸探针的标记方法地高辛标记核酸探针的标记方法核酸探针已被广泛用于筛选重组克隆、基因多样性的种性检测和真菌种群内及种群之间的系统发育关系评价。
最早使用的放射性同位素标记核酸探针具有敏感性高、特异性好、分辨力强的特点,但放射性同位素标记也存在着一系列令人困扰的问题,如成本高、探针半衰期短、放射性物质危害人体健康等。
而且在进行放射性同位素标记实验时,需要有专门的实验室及相应的实验保护设施,还需要由经过培训的专业人员来操作,因而限制了在普通实验室进行分子生物学实验。
近几年发展起来的非放射性核酸探针大多通过酶促、光化学和化学手段掺入一种报道基团,这种报道基团可通过高灵敏度的冷光、荧光或金属沉淀等检测系统检测。
另外,应用pH电极或感应器技术的电化学检测系统也有报道。
在这些检测系统中,灵敏度最高的是生物素- 亲合素检测系统和半抗原-抗半抗原地高辛检测系统。
由于生物样品中常含有内源性生物素及生物结合蛋白,生物素标记的核酸探针会发生一些非特异性结合,从而影响实验效果。
与生物素-亲合素系统同样具有高灵敏度,却减少了非特异性结合的地高辛检测系统,已为人们所接受,并得到广泛的应用。
地高辛(Digoxigenin ,DIG) 又称异羟基洋地黄毒甙元,是一种类固醇半抗原分子。
其化学结构如图1 所示。
采用人工方法可以将地高辛的线型间隔臂与dUTP 连接起来,形成DIG-11-dUTP,通过随机引物法或PCR法将其掺入到DNA探针中。
RNA探针的标记是使用噬菌体信息编码的RNA聚合酶,通过体外转录将DIG-11-dUTP掺入到RNA探针中。
寡核苷酸探针的标记则是通过末端转移酶催化,在3,末端加上DIG-11-dUTP/dATP 或DIG-11-ddUTP 尾巴。
对于目的DNA 或RNA 来说,分子杂交后,杂交部分可通过ELISA 实验程序加以检测,即加入一种结合有碱性磷酸酶的地高辛-特异性抗体,它与地高辛半抗原分子形成酶联抗体-半抗原(DIG) 复合物,再加入相应的显色底物,使杂交部分得以显示。
Southern杂交探针标记及检测试剂盒:DIG High Prime DNA Labeling and Detection Starter Kit II (detection with CSPD)原理所谓DNA探针,实质上是一段已知的基因片段,应用这一基因片段即可与待测样品杂交。
如果靶基因和探针的核苷酸序列相同,就可按碱基配对原则进行核酸分子杂交,从而达到检查样品基因的目的。
在随机引物法标记反应液中,有随机合成的六聚体核昔酸(hexanucleotide)作为引物,dATP、dCTP、dGTP、dTTP 和DIG-dUTP作为合成底物,以单链DNA作为模板,在Klenow酶的作用下,合成掺入地高辛的DNA链。
以地高辛标记的探针与靶基因DNA链杂交后,再通过免疫反应来进行检测。
一般通过酶标抗地高辛抗体来检测,就可以肯定杂交反应的存在。
免疫检测采用CSPD发光曝光到X光片。
试剂盒内容:1. DIG-High Prime,vial 1,50ul(5×);2. 地高辛标记的对照DNA,vial 2,20ul(5ug/ml pBR328 DNA);3. DNA稀释缓冲液,vial 3,(3×1ml);4. AP Conjugate,vial 4,50ul(750U/ml);开封后2-8℃稳定保存。
5. CSPD,vial 5,50 ml;开封后2-8℃避光稳定保存。
6. Blocking solution(封闭液),vial 6,4×100ml(10×);开封后应分装,在-15~-25℃稳定保存,或2-8℃保存1个月,工作液应现用现配。
7. DIG Easy Hyb Granules,vial 7,4×100ml。
表1 需准备的其他试剂及设备过程设备试剂DIG标记探针水浴、电炉双蒸水,灭菌;0.2 M EDTA溶液(pH 8.0),灭菌。
探针灵敏度检测尼龙膜(Hybond-N+ NylonMembrane)洗脱缓冲液马来酸缓冲液检测缓冲液转膜转膜装置、紫外交联装置或烘箱TBE,酸变性液,碱变性液,中和液,20×SSC杂交电炉、冰水混合物、尼龙膜、杂交炉、杂交瓶20×SSC,1% SDS免疫检测洗脱缓冲液;马来酸缓冲液;检测缓冲液;重新杂交水浴20×SSC;1% SDS;1 M NaOH试剂配制:1. 马来酸缓冲液——0.1 M马来酸(M.W. 116.1),0.15 M NaCl,NaOH固体调pH至7.5,常温(称取11.61g马来酸,8.77g NaCl,定容至1000mL,高压灭菌)。
Southern印迹杂交实验原理和方法-3真空转移法的最大优点是迅速,可在转膜的同时进行DNA变性与中和整个过程约需3 0~60分钟。
但在操作中应注意两个问题,一是真空压力不能太大,若压力过大,凝胶被压缩,转移效率会降低;二是真空转移液要密封严,防止漏气影响压力的产生。
下表列出了不同的印迹方法。
表10-2 不同印迹方法的比较表五、探针标记用于Southern印迹杂交的探针可以是纯化的DNA片段或寡核苷酸片段。
探针可以用放射性物质标记或用地高辛标记,放射性标记灵敏度高,效果好;地高辛标记没有半衰期,安全性好。
人工合成的短寡核苷酸可以用T4 多聚核苷酸激酶进行末端标记。
探针标记的方法有随机引物法、切口平移法和末端标记法,详细方法参见本书相关章节。
这里介绍放射标记。
以下为 Promega公司随机引物试剂盒提供的标记步骤:(一)取25~50mg模板DNA于0.5ml离心管中,100℃水浴5min,立即置冰浴。
(二)在另一个0.5ml离心管中加入:Labeling 5×Buffer (含随机引物) 10μldNTPmix(含dCTP.dGTP.dTTP各0.5mmol/L) 2μlBSA(小牛血清蛋白) 2μl[α-32ρ] dATP 3μlKlenow 酶 5U(三)将变性模板DNA加入到上管中,加ddH2O至50μl混匀。
室温或37℃ 1h.(四)加50μl终止缓冲液终止反应标记后的探针可直接使用或过柱纯化后使用。
由于α-32ρ的半衰期只有14天,所以标记好的探针应尽快使用。
探针的比活性最好大于 1091计数/分/μl。
六、预杂交(prehybridizafion)将固定于膜上的DNA片段与探针进行杂交之前,必须先进行一个预杂交的过程。
因为能结合DNA片段的膜同样能够结合探针DNA,在进行杂交前,必须将膜上所有能与DNA结合的位点全部封闭,这就是预杂交的目的。
预杂交是将转印后的滤膜置于一个浸泡在水浴摇床的封闭塑料袋中进行,袋中装有预杂交液,使预杂交液不断在膜上流动。
地高辛杂交试剂盒:DIG Northern Starter Kit (ROCHE) 12039672910原理:RNA是通过异羟基洋地黄毒苷(digoxigenin,Dig)配基标记的脱氧尿嘧啶核苷三磷酸(dUTP)随机插入结合而被标记。
dUTP通过间臂连结类固醇半抗原异羟基洋地黄毒苷酸基,形成Dig-dUTP,杂交反应后,杂交的靶RNA通过酶联免疫法与一个抗体复合物抗异羟基洋地黄毒苷配基碱性磷酸酶复合物(Dig)Ap结合,接着在5-溴-4氯-3-吲哚磷酸盐(X-磷酸盐)和硝基四氮唑蓝(NBT)存在下,由酶催化反应,在杂交部位形成蓝紫色带或颗粒;也可以使用荧光碱性磷酸酶底物,检测的灵敏度常规可达到0.1pg.探针制备(1)模板引物设计,将T7 RNA polymerase序列5′-TAATACGACTCACTATAGGG-3′加在目的基因下游引物前面。
模板的准备,先摸索模板的率高的PCR体系,25μL的体系扩增20管。
PCR产物纯化1)将20管的PCR产物转移入1.5mL离心管加入等体积的氯仿:异丙醇(24:1),抽提一次,12000rpm,离心,10min(此步上清中可能会有些浑浊,但没关系)。
2)上清转移入一个新的1.5mL 离心管中,并向其加入1/10倍体积的NaAc,2-2.5倍体积的无水乙醇,-20℃沉淀至少3h或过夜沉淀12000rpm,离心,15min。
3)用75%的酒精洗涤沉淀一次,50µL的DEPC处理水回溶。
4)用中孔点样,89V电泳,用质量比较好的胶回收试剂盒进行胶回收,30µL的DEPC处理水回溶,用分光光度计测DNA 的浓度,最好是在200ng/µL左右。
(2)取一只经过DEPC处理并高压灭菌的无RNase污染的1.5mL离心管中依次加入下列反应物(离心管置于冰上)。
模板DNA (200ng)混合物5×(1a) 2 μL缓冲液5×(1b) 2 μL聚合酶T7 (3号管) 1 μLddH2O Up to 10 μL模板DNA一定不能多于200ng,量比较多,合成探针的效率较低,此步骤非常关键;最好1μLDNA 的浓度为200ng,剩余补DEPC水。
地高辛标记探针在Sou thern杂交分析中的技术要点陆小平 周文军(苏州大学生命科学学院江苏苏州215006)小岛峰雄(日本信州大学纤维学部) 外源基因是否成功导入受体材料的基因组中,必须从转化植株中找到分子生物学的检测证据。
目前,PCR、Southern杂交等作为常用检测手段而被广泛采用。
虽然PCR技术可以快速得到结果,但是,以农杆菌介导的材料必须慎重这一结论,以免由于农杆菌污染造成假阳性。
而Southern分析由于操作程序繁琐,对植物基因组DNA的提取、纯化、酶切等技术要求较高,有时使杂交结果不甚理想。
我们在植物基因转化的研究中,对荞麦,桑树,紫景天,洋麻等基因组DNA的提取、纯化、酶切进行了探讨,对流程中的有关步骤进行技术改良,使酶切后的PNA在凝胶板上是涂布状完全达到了Southern杂交的技术要求,并用地高辛(D ig)标记探针对外源DNA进行分析,取到了较好的效果。
现简述如下∶1 植物基因组D NA的提取及纯化1)提取高纯度的DNA是Southern杂交的关键, DNA的粗提物中,往往含有大量的蛋白质、多糖、单宁、色素等大分子杂质。
这些杂质通常与DNA共同沉淀或与DNA聚合成大分子复合物。
一旦复合物形成,即便在以后的操作中用酚、氯仿多次纯化,也很难将其除去。
我们的经验是:当用液氮破碎新鲜样品的细胞壁后,先用蒸馏水洗脱两次(洗脱温度为42℃),每次5m in。
以达到洗脱多糖的目的。
每次洗脱后离心5m in(13000 r m in),弃去上层液。
该上层液中含有粘度较高的胶状体,其主要成分可能是粘性多糖。
在提取桑树基因组DNA时,最好用叶柄或幼茎、幼叶作提取材料。
在样品有限时,也可用成熟叶甚至老叶代替。
据C lark M S (1998)介绍,取样前对材料进行除淀粉处理(减少光照、黑暗处理24h),可以抑制多糖的污染。
但我们采用除淀粉处理后的实验结果并不理想,其效果远不及用蒸馏水洗脱。
另外,该方法不仅可以用于桑树DNA提取,而且也适用于其他植物材料(果树、花卉)。
2)由于桑叶中也含有酚类、色素,提取DNA时,常有茶褐色物质混入DNA中,由此而影响DNA的纯度。
为了防止DNA的褐变,我们在液氮磨碎后,立即置冰箱下层或4℃条件下任其自然解冻,待粉末完全解冻后再加入DNA提取液。
3)做一次Southern杂交所需10Λg纯DNA,一般取0.2g新鲜样品即可得到10Λg以上纯DNA。
因此,用该方法提取DNA无论是产量还是质量都能满足一定要求。
为了保证DNA的纯度,每管加样量不要太多,以0.2~0.25g新鲜样品为宜,用30mL液氮研磨成粉末,置4℃条件下解冻后分别加600ΛL提取液 和200ΛL提取液 ,再稍稍研磨后全部转入2mL的离心管中,其余步骤仍按试剂盒要求操作。
4)在去除蛋白质的干扰时,我们仍用蛋白酶K,但其中添加了80ΛL酶解缓冲液(60mmo l T h is2HC l、60mmo l ED TA、3%SD S、pH7.8),置55℃条件下酶切过夜。
结束后再用苯酚、氯仿处理。
5)纯化后的DNA可以通过测定波长为230、260、280的紫外吸收光谱来确定其浓度,但这些数据只能作为参考,因即便有较高的OD值,仍会存有影响酶切的干扰物质,我们建议最好采用凝胶电泳检测。
2 酶切DNA的酶切时间一般是1~3h,但结束前最好取10ΛL(总量为200ΛL)酶切产物在小孔胶上检测是否完全酶切,即DNA片段弥散于各泳道中,若加样孔下方仍有亮度较强的条带出现(重复序列例外),这表明DNA尚未完全酶切,需继续延长酶切时间。
3 Southern印迹将完全酶切的DNA上大孔胶电泳,若DNA在胶板上呈涂布状,便可进行Southern印迹。
若近加样孔一侧的泳带,其前沿参差不齐;加样孔变形;孔内残留物较多;泳道中DNA分布不匀等,这些均为干扰物存在所致。
在条件许可时应重新酶切。
Southern印迹可按常规方法操作,将胶板分别用0.25mo l HC l、变性液、中和液浸泡15~30m in后,用20XSSC溶液将变性DNA全部转移至尼龙膜(H ybo rdN+m em bane)上。
但印迹操作时,须戴手套作业,以免污染尼龙膜而影响DNA的固定。
吸水纸上的重量要逐次添加,以防泳道变形和凝胶毛细管过早堵塞。
4 杂交1)固定 印迹结束后,取下尼龙膜,置室温中自然干燥1h,用紫外仪[FUNA2UV L IKER(FS21500)]将DNA固定到尼龙膜上(约30s)。
2)预杂交 将尼龙膜装入小塑料袋中,加入10mL 预杂交液后,驱尽袋中的气泡,封口后置65℃杂交炉(EYELA H YBR I D Z A T I ON OV EN M H S2301)中孵育1h。
3)杂交 剪开小塑料袋的一角,直接加入10ΛL变性D ig探针,赶尽气泡后重新封口,继续置65℃杂交炉中慢速振摇过夜。
该步骤的关键是小塑料袋中不能留有气泡,否则,果蝇唾腺染色体的几种染色方法比较郜 刚(山西师范大学生命科学学院山西临汾041004)《生物学通报》编委:您好。
笔者对贵刊2002年第37卷第2期第23页刊登的“用Feuglen染色法制作果蝇唾腺染色体”一文感兴趣,但笔者认为文中几处不妥,比如1)水浴温度波幅偏高;2)染色时间不确定;3)试剂配方不全;4)盐酸的浓度单位有误,mo l?M?;5)注意事项影响实验结果表述不清。
特撰写下文与各位读者商榷。
双翅类昆虫如黑腹果蝇(D rosop h ila m elanog aster)的唾腺染色体(Salivary ch romo som e)比普通染色体大的多,处于体细胞同源染色体的配对状态,是由于唾腺染色体经过多次复制而并不分开形成的,大约有1000~4000根染色体丝的拷贝,故又称为多线染色体(Po lytene ch romo som e)。
它是观察染色体形态、研究染色体结构变异等的好材料。
制作果蝇唾腺染色体标本的染色方法一般有3种:醋酸洋红法、苯酚品红法和孚尔根(Feuglen)染色法(除此之外还有其他方法)。
各种方法都有其自身的特点及适用的条件,因此没有1种染色方法是普遍适用完美无缺的。
现将3种常用方法的优缺点分述如下,并提出1个实用的永久封片制作方法。
1 醋酸洋红法洋红的常用浓度为0.5%~1.0%,醋酸常用浓度为45%~50%,一般现配现用较好。
洋红是从胭脂虫(Coccus cacti)的雌虫中提取的作为染料的提取物,提取物的品质因胭脂虫的种类而异,是一种混合物,其中具有染色活性的是洋红酸。
洋红酸是一种二元弱酸,如果溶于碱性溶液中,则具有酸性染料的性质,可使细胞质着色;如果溶于酸性溶液中,则具有碱性染料的性质,可使染色质(体)着色。
此法多用滴染法,快速简便,为改进其染色效果,也可采用浸染法,并辅以火焰微热(即滴加醋酸洋红盖片后在酒精灯火焰上微热),增加本底清晰度,加大反差。
醋酸洋红的配制和染色都比较简单,对细胞穿透力较强,这是其主要优点,此外它对染色体和核仁均可染色,故也适用于减数分裂的细胞染色。
但其染色强度和分色效果不及其他染色剂,通常只作临时染色观察,不用于制作永久性装片。
也可以用醋酸地衣红替代洋红,这样细胞质着色较少,效果较好。
2 孚尔根染色法孚尔根染色法是常用于鉴别细胞中DNA的一种组织化学方法,细胞核经过温和的盐酸的水解作用杂交信号将被冲散。
加探针时不要碰到尼龙膜,以免膜与高浓度探针发生特异性结合而出现污染斑点。
5 洗膜取出尼龙膜,置漂洗液(2X、1X、0.1X SSPE 0. 1%SD S)中逐级漂洗10~15m in,以洗脱未杂交的D ig 探针和非特异性结合。
6 显影将尼龙膜按下列程序再次浸洗:1)80mL缓冲液 (0.1mo l马来酸 0.15mo l氯化钠 pH7.5)10m in;2)80mL缓冲液 (72mL缓冲液 8mL10%阻断剂B locling Roaget)60m in;3)20mL缓冲液 (20mL缓冲液 3U A nti2D ig2A P)30m in;4)80mL缓冲液 2次,每次10m in;5)20mL缓冲液 (0.1mo l氯化钠 0.1mo l T ris2HC l 50mmo l氯化镁 pH9.5)5m in;6)20mL显影液(3.76m g磷酸溴氯吲哚 7.5m g 氯化氮蓝四唑 20mL蒸馏水)黑暗中3~6h。
7 膜的保存杂交信号在3h可达到高峰,并可稳定24h。
若信号过强,可缩短显影时间,当信号强度达到要求后,即可停止显影。
保存时取出尼龙膜,用T E漂洗2次后封膜保存。
膜干燥或久置后,信号将有所减弱或褪色,但经T E湿润后仍会重现原有的杂交信号。
用32P标记探针分析转化株中的外源DNA是分子杂交的常用手段,但32P保存时间有限,对操作人员有一定辐射,废液对环境污染严重,而D ig探针可以弥补这些不足。
因D ig标记属生物素类,其主要成分(洋地黄毒苷配基)是由植物洋地黄中提取的半抗原类固醇物质,经PCR反应制作的探针没有辐射污染;-20℃中可保存12个月;杂交液可重复使用2~3次;敏感性高、检测速度快。
因此,目前世界一些研究组织提倡用地高辛代替32P标记。
参考文献 1 C lark M S主编顾红雅瞿礼嘉主译.植物分子生物学实验手册.北京:高等教育出版社,1998. 2 陈丙莺,陈子兴主编.分子生物学基础与临床,南京:东南大学出版社,2000,2. 3 王关林,方宏筠主编.植物基因工程原理与技术.北京:科学出版社,1998.(BH) 。