探针的标记及产物的纯化
- 格式:ppt
- 大小:57.00 KB
- 文档页数:14

Northern Blot实验方案一.探针准备—体外转录1.RNA探针的制备基本要求:不被RNase污染。
(1)制备线性DNA模板a.引物设计采用Array Designer 4设计引物,在下游引物的5'端加上T7启动子序列(蓝色部分标示)。
如下例:AGTAATTGGAATTACAGCT AGGCAGCAAGCGAATGAATCCCGATA AGCTGACTCACGTCAGTGATTCGGGTGAGTGAGTGCCGCTAACAA CGCTGCAATTTCAAGTAAGAAGAGTGTTACCCAAAGTAATTGGAA TTACAGCTAGGCAGCAAGCGAATGAATCCCGATAAGCTGACTCAC GTCAGTGATTCGGGTGAGTGAGTGCCGCTAACAACGC TGCAATTT CAAGTAAGAAGGGTGTTACCCAAAGTAATTGGAATTACAGCTAGG CAGCAAGCGAATGAATCCCGATGAGCTGACTCACGTCAGTGATTC GGGTGAGTGAGTGCCGCTAACAACGCTGCAATTTCAAGTAAGAAG Probe-F: 5'AGGCAGCAAGCGAATGAATC3'probe-R:5'AATTGTAATACGACTCACTATAGGGCG GCGTTGTTAGCGGCACTC3' 预期产物大小:198bp引物设计原则同一般引物设计原则。
Array Designer 4可自行评价引物质量,选取引物时选best即可。
b.探针模板的的制备。
以91001染色体为模板,PCR扩增获得探针模板。
普通琼脂糖凝胶电泳检测模板大小是否正确。
扩增产物的纯化:产物用QIAquick PCR purification Kit(QIAGEN)纯化。
纯化后用DEPC水溶解,测浓度。
—20℃冻存。
(2)体外转录反应制备探针(20 µl)两种方法方法一:a.NTPs 的配置:10×NTPs:ATP/GTP/CTP 各5µl(100mM)UTP 3.25µl(100mM)DMPC处理水31.75µl充分混匀,-20℃保存。

1、准备工作:A、取出TdT Buffer (5X)、Biotin-11-dUTP和Ultrapure water溶解,并置于冰浴上备用。
B、取出待标记的单链EMSA探针,用水稀释至1μM,并置于冰浴上备用。
如果待标记的EMSA探针为双链,95℃加热2分钟,然后立即放置到冰水浴中,使双链的EMSA探针转变为单链的探针,然后同样用水稀释至总的单链DNA浓度为1μM,即每条单链的浓度为0.5μM,相当于最初双链的EMSA 探针浓度为0.5μM。
2、DNA探针的标记:Ultrapure water 29μlTdT Buffer(5X) 10μl待标记探针(1μM) 5μlBiotin-11-dUTP(5μM) 5μlTdT(10U/μl) 1μl总体积50μlA、参考上述设置反应体系。
注:对于双链的EMSA探针的标记反应,建议一次做两管,即总体积共100μl,以最终获得足够的生物素标记EMSA探针用于后续EMSA检测。
B、用枪轻轻吹打混匀,切勿vortex。
37℃孵育30分钟。
C、加入2.5μl 探针标记终止液,轻轻混匀终止反应。
3、TdT的去除:A、探针标记反应终止后,加入52.5μl氯仿-异戊醇(24:1),vortex使有机相和水相充分混合以抽提TdT(说明:静止后有机相和水相会很快分层)。
B、12000-14000g离心1-2分钟。
吸取上清备用。
上清即为被生物素标记的单链DNA探针。
4、探针的纯化(选做):通常为实验简便起见,可以不必纯化标记好的探针。
有些时候,纯化后的探针会改善后续实验的结果。
如需纯化,可以按照如下步骤操作:A、对于100μl标记好的探针,加入1/4体积即25μl的5M醋酸铵,再加入2体积即200μl的无水乙醇,混匀。
B、-70℃至-80℃沉淀1小时,或-20℃沉淀过夜。
C、4℃,12,000g-16,000g离心30分钟。
小心去除上清,切不可触及沉淀。
D、4℃,12,000g-16,000g离心1分钟。
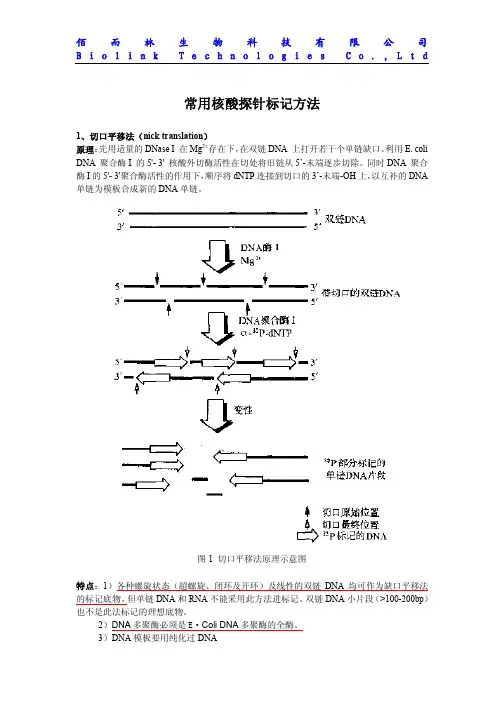
常用核酸探针标记方法1、切口平移法(nick translation)原理:先用适量的DNase I 在Mg2+存在下,在双链DNA 上打开若干个单链缺口。
利用E. coli DNA 聚合酶I 的5'- 3' 核酸外切酶活性在切处将旧链从5’-末端逐步切除。
同时DNA 聚合酶I的5'- 3'聚合酶活性的作用下,顺序将dNTP连接到切口的3’-末端-OH上,以互补的DNA 单链为模板合成新的DNA单链。
图1 切口平移法原理示意图特点:1)各种螺旋状态(超螺旋、闭环及开环)及线性的双链DNA均可作为缺口平移法的标记底物。
但单链DNA和RNA不能采用此方法进标记。
双链DNA小片段(>100-200bp)也不是此法标记的理想底物。
2)DNA多聚酶必须是E·Coli DNA多聚酶的全酶。
3)DNA模板要用纯化过DNA2、随机引物法(random priming)原理:将待标记的DNA探针片段变性后与随机引物一起杂交,然后以此杂交的寡核苷酸为引物,大肠杆菌DNA聚合酶I大片段(E·Coli DNA polymerase I Klenow Fragment)的催化下,合成与探针DNA互补的DNA链。
当反应液中含有标记的dNTP时,即形成标记的探针。
图2 随机引物标记法原理示意图特点:1)除了能进行双链DNA标记外,也可用于单链DNA和RNA探针的标记。
2)所得到的标记产物是新合成的DNA单链,而所加入的DNA片段本身并不能被标记。
3)新形成的标记DNA单链的长度与加入寡核苷酸引物的量成反比,因为加入的寡核苷酸数量越多,合成起点也越多,得到的片段的长度也越短。
按标准方法得到的标记产物长度一般为200-400bp。
3、末端标记与切口平移法和随机引物法不同,DNA末端标记法并不将DNA片段的全长进行标记,而是只将其一端(5’或3’端)进行部分标记。
其特点是可得到全长DNA片段,DNA片段并非均匀标记,标记活性不高。

生物素标记探针合成引言:生物素标记探针是生物学研究中常用的实验工具之一,通过将生物素与待标记的分子(如蛋白质、核酸等)结合,可以实现对目标分子的检测、定位和追踪等功能。
本文将介绍生物素标记探针的合成方法及其在生命科学研究中的应用。
一、生物素标记探针的合成方法1.1 生物素的化学合成生物素是一种含有硫辛烷环的小分子有机物,可以通过化学合成的方法得到。
一种常用的合成方法是利用二巯基乙酸(DTNB)与己二酸缩合反应,得到二巯基生物素。
然后,通过脱保护反应和硫辛烷环的形成,最终得到生物素。
该方法合成的生物素纯度高,适用于大规模合成。
1.2 生物素的修饰生物素通常需要在特定位置引入修饰基团,以便与待标记的分子结合。
修饰方法有多种,常见的包括化学修饰和生物修饰两种。
化学修饰方法包括使用活化的酯化试剂将生物素与分子中的氨基、羟基等官能团连接起来,形成酯键。
这种方法简单易行,适用于多种分子的修饰。
生物修饰方法则利用生物素连接酶(biotin ligase),通过生物素连接酶与特定的肽标签结合,实现生物素的共价连接。
这种方法具有高度特异性,适用于对特定肽链进行修饰。
二、生物素标记探针的应用2.1 免疫组化免疫组化是一种常用的细胞和组织学研究方法,通过对生物样本中的目标分子进行特异性标记,实现对其在细胞或组织中的定位和表达水平的检测。
生物素标记的抗体是免疫组化实验中常用的标记物之一。
通过将生物素标记的抗体与细胞或组织样本中的目标分子结合,再利用辣根过氧化物酶-生物素-抗生素酶(HRP-biotin-avidin)体系,可实现对目标分子的高灵敏度检测。
2.2 蛋白质互作研究生物素标记探针在蛋白质互作研究中发挥着重要作用。
通过将生物素标记的蛋白质与待检测的蛋白质样本反应,再利用亲合纯化技术,如亲和层析、免疫沉淀等,可以富集目标蛋白质及其相互作用的复合物。
接着,通过质谱分析等方法,可以鉴定和分析复合物中的蛋白质成分,进而揭示蛋白质互作网络和信号通路。

原位杂交原理及具体操作原位杂交的具体操作包括以下几个步骤:1.样本准备:收集需要研究的细胞或组织样品,并进行固定和处理。
具体处理方法根据研究对象的特点和需求而定,可以涉及蛋白酶消化、脱脂、固定等步骤。
2.产生标记探针:首先要选择合适的探针来检测目标序列。
探针可以是DNA或RNA序列,根据研究对象选择相应的方法进行标记,常见的标记方法有荧光标记、放射性标记和酶标记等。
标记后的探针需要经过纯化和检测,确保标记效果良好。
3.杂交反应:将标记的探针与样本中的DNA或RNA进行杂交反应。
首先需要将样本脱水,并在适当温度下使用探针溶液进行孵育反应,使探针与目标序列发生特异性结合。
杂交温度根据探针和目标序列的互补性来确定,一般在适当的温度下进行持续反应。
4.洗涤:杂交反应结束后,需要进行严格的洗涤步骤,以去除未结合的探针和非特异性结合。
洗涤的条件也根据研究需要而定,可以使用高盐溶液、低盐溶液或有机溶剂等来去除非特异性结合。
5.信号检测:根据具体标记方法的不同,可以使用荧光显微镜、射线计数器或底物染色等方法来检测标记的探针。
通过观察探针的位置和强度,可以得到目标序列在细胞或组织中的分布和表达情况。
6.分析与图像处理:根据实验结果,可以对图像进行定量分析和处理。
现代技术已经能够通过图像软件进行分析和定量,得到原位杂交的定量数据。
原位杂交技术的优点在于可以在细胞或组织水平上观察和定位目标序列的存在和表达情况,为研究基因表达、基因功能以及病理学等提供了强有力的工具。
但是在操作过程中需要注意探针的选择和合成、杂交条件的优化以及样品处理的标准化等问题,以确保实验结果的准确性和可重复性。

地高辛标记核酸探针的标记方法Last revision on 21 December 2020地高辛标记核酸探针的标记方法核酸探针已被广泛用于筛选重组克隆、基因多样性的种性检测和真菌种群内及种群之间的系统发育关系评价。
最早使用的放射性同位素标记核酸探针具有敏感性高、特异性好、分辨力强的特点,但放射性同位素标记也存在着一系列令人困扰的问题,如成本高、探针半衰期短、放射性物质危害人体健康等。
而且在进行放射性同位素标记实验时,需要有专门的实验室及相应的实验保护设施,还需要由经过培训的专业人员来操作,因而限制了在普通实验室进行分子生物学实验。
近几年发展起来的非放射性核酸探针大多通过酶促、光化学和化学手段掺入一种报道基团,这种报道基团可通过高灵敏度的冷光、荧光或金属沉淀等检测系统检测。
另外,应用pH电极或感应器技术的电化学检测系统也有报道。
在这些检测系统中,灵敏度最高的是生物素- 亲合素检测系统和半抗原-抗半抗原地高辛检测系统。
由于生物样品中常含有内源性生物素及生物结合蛋白,生物素标记的核酸探针会发生一些非特异性结合,从而影响实验效果。
与生物素-亲合素系统同样具有高灵敏度,却减少了非特异性结合的地高辛检测系统,已为人们所接受,并得到广泛的应用。
地高辛(Digoxigenin ,DIG) 又称异羟基洋地黄毒甙元,是一种类固醇半抗原分子。
其化学结构如图1 所示。
采用人工方法可以将地高辛的线型间隔臂与dUTP 连接起来,形成DIG-11-dUTP,通过随机引物法或PCR法将其掺入到DNA探针中。
RNA探针的标记是使用噬菌体信息编码的RNA聚合酶,通过体外转录将DIG-11-dUTP掺入到RNA探针中。
寡核苷酸探针的标记则是通过末端转移酶催化,在3'末端加上DIG-11-dUTP/dATP 或DIG-11-ddUTP 尾巴。
对于目的DNA 或RNA 来说,分子杂交后,杂交部分可通过ELISA 实验程序加以检测,即加入一种结合有碱性磷酸酶的地高辛-特异性抗体,它与地高辛半抗原分子形成酶联抗体-半抗原(DIG) 复合物,再加入相应的显色底物,使杂交部分得以显示。

Electrophoretic Mobility Shift Assay--电泳迁移率检测点击次数:137 发表于:2008-06-18 14:53转载请注明来自丁香园来源:互联网凝胶迁移或电泳迁移率检测(Electrophoretic Mobility Shift Assay,EMSA)是一种检测蛋白质和DNA序列相互结合的技术,最初用于研究DNA结合蛋白和其相关的DNA结合序列相互作用,可用于定性和定量分析。
这一技术目前已用于研究RNA结合蛋白和特定的RNA序列的相互作用,已经成为转录因子研究的经典方法。
其基本原理是蛋白质可以与末端标记的核酸探针结合,电泳时这种复合物比无蛋白结合的探针在凝胶中泳动的速度慢,即表现为相对滞后。
该方法可用于检测DNA结合蛋白、RNA结合蛋白,并可通过加入特异性的抗体(supershift)来检测特定的蛋白质,并可进行未知蛋白的鉴定。
生物体内DNA转录成RNA是基因表达的关键过程,基因表达调控主要发生在转录水平。
近年来,基因分子生物学研究领域的趋势之一是逐渐从基因结构和功能分析转到基因顺式作用元件和转录因子及其转录调控机理上,对基因转录调控的研究将是今后相当长一段时期内功能基因组学研究的热点之一。
在转录水平,基因的转录行为是由顺式作用元件(cis-acting elements)和反式作用因子(trans-acting factors)(即转录因子)相互作用调控的,因此要探讨基因的表达调控规律,分离、鉴定基因的位点控制区(loci control region,LCR)中顺式作用元件和相应转录因子至关重要。
但仅仅如此还不够,对于基因表达调控,必须从三个层次展开:一是分离和鉴定基因5'端核心启动子等顺式作用元件,二是分离和鉴定与各顺式作用元件相对应的转录因子,三是检测各顺式作用元件与对应转录因子的相互作用。
鉴定顺式调控元件和转录因子是研究得比较多的内容,而对于顺式作用元件与对应转录因子的相互作用这个层次的研究相对较为薄弱。


凝胶迁移或电泳迁移率检测(Electrophoretic Mobility Shift Assay, EMSA)是一种检测蛋白质和DNA序列相互结合的技术,最初用于研究DNA结合蛋白和其相关的DNA结合序列相互作用,可用于定性和定量分析。
这一技术目前已用于研究RNA结合蛋白和特定的RNA序列的相互作用。
由于很多研究的TF不具有结合的特异性,所以即使发现TF和被研究的寡核苷酸探针结合,也不表示在体内该TF不能和其它寡核苷酸结合,运用一些生物信息学软件,可以模拟表示TF与基因启动子区寡核苷酸结合的具体情况,发现TF可以和启动子区PUTATIVE 结合。
同时,由于EMSA在体外不能模拟细胞内众多生物分子的相互作用,比如,某一TF在细胞内由于受到其它TF的协同或者修饰后才能与寡核苷酸结合的话,那么在体外是不能重现这一结果的。
03 年上一篇关于ChIP方法的介绍文献攻击EMSA 没有考虑细胞内完整自然的染色质结构和调节的影响而受到很大的限制!下面是在一个PROTOCAL1.探针的标记:(1) 如下设置探针标记的反应体系:待标记探针(1.75pmol/微升) 2微升T4 Polynucleotide Kinase Buffer (10X) 1微升Nuclease-Free Water 5微升[γ-32P]A TP(3,000Ci/mmol at 10mCi/ml) 1微升T4 Polynucleotide Kinase (5-10u/微升) 1微升总体积10微升按照上述反应体系依次加入各种试剂,加入同位素后,V ortex混匀,再加入T4 Polynucleotide Kinase,混匀。
(2) 使用水浴或PCR仪,37℃反应10分钟。
(3) 加入1微升探针标记终止液,混匀,终止探针标记反应。
(4) 再加入89微升TE,混匀。
此时可以取少量探针用于检测标记的效率。
通常标记的效率在30%以上,即总放射性的30%以上标记到了探针上。

基因探针检测过程
基因探针(Gene Probe)检测是一种分子生物学技术,用于检测特定基因或DNA序列的存在和表达。
下面是基因探针检测的一般过程:
1.制备探针:
•选择目标基因或DNA序列:确定要检测的目标基因或DNA序列。
•合成或标记探针:探针可以通过合成DNA序列或标记已有的DNA序列制备。
标记通常使用放射性同位素(如32P或
35S)或非放射性标记物(如荧光染料、酶等)。
2.制备样品:
•提取DNA:从待测样本中提取目标DNA。
样本可以是血液、细胞、组织等。
•切割DNA:使用特定的酶酶解DNA,将其切割成适当的片段。
3.杂交反应:
•杂交液:将制备好的探针与目标DNA混合在一起,形成杂交液。
•条件设定:设定适当的温度和时间,使探针与目标DNA发生杂交。
这一步可以通过加热和冷却过程来模拟DNA的分离和
结合。
4.检测信号:
•放射性同位素标记:如果使用放射性同位素标记的探针,
通过暗室摄影或闪烁计数器等设备检测辐射信号。
•非放射性标记:如果使用非放射性标记物,比如荧光标记,通过荧光显微镜或流式细胞仪等设备检测信号。
5.结果分析:
•解释结果:根据信号的强弱和位置,确定目标基因或DNA 序列是否存在。
•结果验证:可以通过其他技术手段如聚合酶链反应
(PCR)、Southern印迹等来验证基因探针检测的结果。
基因探针检测是一种敏感、特异、高效的分子生物学技术,广泛应用于医学、生物学研究和疾病诊断等领域。

标记探针制备
标记探针制备方法是一种用于生物学和医学研究的重要技术,它使用标记探针(例如核酸、碳水化合物或蛋白质)来克隆、检测和定位特定的DNA序列或蛋白质。
该技术在遗传学、分子生物学、转基因研究、微生物学和药物发现等领域有着广泛的应用。
标记探针制备的过程主要包括几个步骤:
1. 选择目标序列:首先,研究者需要选择他们想要克隆或检测的特定DNA序列或蛋白质序列作为标记探针的目标序列。
这可以通过搜索数据库或通过任何其他方法实现。
2. 合成标记探针:接下来,研究者可以使用任何一种合成技术(例如PCR、合成DNA或全基因组合成)来合成标记探针,以实现对特定序列的克隆和检测。
3. 加入标记物:研究者可以使用任何一种方法将标记物(如核酸、荧光素或放射性同位素)加入到已经合成的标记探针中,以便将其与特定序列区分开来。
4. 纯化探针:最后,研究者可以使用纯化技术将标记探针从其他成分中纯化出来,以便用于实验。
标记探针制备技术的使用有助于研究者更好地深入理解和研究人类基因组的结构和功能,也有助于研究者更
快、更准确地确定他们感兴趣的物种的基因组结构,从而为药物发现和基因治疗等应用提供更多有用的信息。
基因探针的原理
基因探针是一种分子生物学技术,可以用来检测DNA序列的存在和表达。
它主要是利用与特定序列相互补的探针来识别样品中的DNA序列。
具体的原理如下:
1. 靶标分离
在进行基因探针试验前,需要从样品中提取目标DNA序列。
这通常通
过化学或物理的方法进行离子或分子分离,以纯化要检测的DNA分子。
2. 探针标记
对于一种探针,可以用荧光、辐射等技术进行标记。
在探针与其目标DNA结合时,标记物会发出信号,从而进行检测。
3. 探针设计
在设计基因探针时,首先需要确定要检测的目标序列。
探针的设计通
常需要考虑探针长度、比特率、配对特异性,以及可能的体外亚硫酸
盐酶促荧光淬灭等问题。
4. 探针验证
在探针设计完成后,需要通过实验验证探针是否可以与目标序列结合,以及探针是否具有特异性和灵敏度。
这通常通过凝胶电泳、PCR扩增、荧光杂交等多种实验手段进行验证。
5. 实验操作
在使用基因探针进行实验时,需要将探针与目标样品进行反应,并通
过探针标记信号的检测来确定是否存在目标DNA序列。
根据实验具体
的流程,可以将探针与目标样品进行荧光染色、杂交等,以便观察和
检测。
总之,基因探针是一种高效、准确的分子生物学技术,可以用于多种
生物学研究和实验操作。
在进行基因探针实验时,需要注意探针的设计、验证和实验操作等多个方面,以确保实验结果的准确性和可靠性。
杭州昊鑫生物科技股份有限公司 htpp://AidQuick PCR Purification KitPCR产物纯化回收试剂盒目录号:DR02适用范围:适用于PCR反应产物、酶切产物DNA片段、探针标记纯化回收,DNA样品浓缩等。
试剂盒组成、储存、稳定性:试剂盒组成保存50次(DR0101)100次(DR0202)200次(DR0203)平衡液室温 5 ml 10 ml 20 ml 结合液BB 室温30 ml 60 ml 100ml漂洗液WB 室温15 ml 25 ml 50 ml 第一次使用前按说明加指定量乙醇洗脱缓冲液EB 室温10 ml 15 ml 20 ml吸附柱EC 室温50个100个200个收集管(2ml)室温50个100个200个本试剂盒在室温储存12个月不影响使用效果。
储存事项:所有的溶液应该是澄清的,如果环境温度低时溶液可能形成沉淀,此时不应该直接使用,可在37℃水浴加热几分钟,即可恢复澄清。
使用前应该恢复到室温储存于低温(4℃或者-20℃)会造成溶液沉淀,影响使用效果,因此运输和储存均在室温下(15℃-25℃)进行。
避免试剂长时间暴露于空气中产生挥发、氧化、pH值变化,各溶液使用后应及时盖紧盖子。
产品介绍:在高离序盐存在的情况下,DNA片断选择性的吸附于离心柱内的硅基质膜上,再通过一系列快速的漂洗-离心的步骤,去蛋白液和漂洗液将引物、核苷酸、蛋白、酶等杂质去除,最后低盐、高pH值的洗脱缓冲液将纯净DNA从硅基质膜上洗脱。
产品特点:1. 离心吸附柱内硅基质膜全部采用进口世界著名公司特制吸附膜,柱与柱之间吸附量差异极小,可重复性好。
克服了国产试剂盒膜质量不稳定的弊端。
2. 使用了优质结合液,不含传统结合液的碘化钠和高氯酸盐,不抑制回收后酶切、连接克隆等下游反应。
3. 结合液加酚红调制成为了黄颜色,便于监测pH值变化从而达到最佳结合效果,大大提高回收效率。
4. 快速、方便,不需要使用有毒的苯酚、氯仿等试剂,也不需要乙醇沉淀。
Northern Blot实验方案一.探针准备—体外转录1.RNA探针的制备基本要求:不被RNase污染。
(1)制备线性DNA模板a.引物设计采用Array Designer 4设计引物,在下游引物的5'端加上T7启动子序列(蓝色部分标示)。
如下例:AGTAATTGGAATTACAGCT AGGCAGCAAGCGAATGAATCCCGATA AGCTGACTCACGTCAGTGATTCGGGTGAGTGAGTGCCGCTAACAA CGCTGCAATTTCAAGTAAGAAGAGTGTTACCCAAAGTAATTGGAA TTACAGCTAGGCAGCAAGCGAATGAATCCCGATAAGCTGACTCAC GTCAGTGATTCGGGTGAGTGAGTGCCGCTAACAACGC TGCAATTT CAAGTAAGAAGGGTGTTACCCAAAGTAATTGGAATTACAGCTAGG CAGCAAGCGAATGAATCCCGATGAGCTGACTCACGTCAGTGATTC GGGTGAGTGAGTGCCGCTAACAACGCTGCAATTTCAAGTAAGAAG Probe-F: 5'AGGCAGCAAGCGAATGAATC3'probe-R:5'AATTGTAATACGACTCACTATAGGGCG GCGTTGTTAGCGGCACTC3' 预期产物大小:198bp引物设计原则同一般引物设计原则。
Array Designer 4可自行评价引物质量,选取引物时选best即可。
b.探针模板的的制备。
以91001染色体为模板,PCR扩增获得探针模板。
普通琼脂糖凝胶电泳检测模板大小是否正确。
扩增产物的纯化:产物用QIAquick PCR purification Kit(QIAGEN)纯化。
纯化后用DEPC水溶解,测浓度。
—20℃冻存。
(2)体外转录反应制备探针(20 µl)两种方法方法一:a.NTPs 的配置:10×NTPs:ATP/GTP/CTP 各5µl(100mM)UTP 3.25µl(100mM)DMPC处理水31.75µl充分混匀,-20℃保存。
taqman探针的引物纯化方法TaqMan探针是一种常用的分子生物学实验技术中的引物。
它通过与待测目标DNA序列靶标的特定区域杂交,并且在荧光探针特殊设计的结构下,可以产生强荧光信号。
因此,在分子生物学研究中,TaqMan探针广泛用于定量PCR反应和基因表达分析。
在使用TaqMan探针之前,首先需要对其进行纯化以去除可能存在的杂质和不纯物。
下面介绍一种常用的TaqMan探针的纯化方法。
首先,从商业来源或合成制备的TaqMan探针可能包含未纯化的杂质,因此需要进行纯化。
一种常见的纯化方法是使用离子交换层析柱。
在实施这种纯化方法之前,我们需要准备一组离子交换层析柱和相关的缓冲液。
首先制备一定浓度的缓冲液,例如磷酸盐缓冲液(pH 7.4),用于样品的加载和洗脱。
接下来,将TaqMan探针样品溶解在缓冲液中,并将其加载到预先平衡好的离子交换柱上。
使用适当的流速和压力,使样品通过柱并与柱填充物中的离子反应。
这样,TaqMan探针可以与柱填充物上的离子发生离子交换,使得探针与杂质分离。
在样品通过之后,用缓冲液进行柱的洗脱,以移除非特异性结合的杂质和离子。
接下来,使用较高浓度或更高pH值的缓冲液,例如甘氨酸缓冲液(pH 8.0),来洗脱TaqMan探针。
最后,将洗脱的纯化产品收集起来,并使用比色法或其他合适的方法来评估其纯度和浓度。
纯化后的TaqMan探针可以用于后续的PCR反应或基因表达分析。
总结而言,使用离子交换层析柱是一种常见且有效的方法来纯化TaqMan探针。
该方法可以帮助去除探针中的杂质和不纯物,从而提高实验的准确性和可靠性。