对氯甲苯的合成
- 格式:doc
- 大小:28.50 KB
- 文档页数:3

第36卷第1期化学反应工程与工艺V ol 36, No 1 2020年2月Chemical Reaction Engineering and Technology Feb. 2020文章编号:1001—7631 ( 2020 ) 01—0060—08DOI: 10.11730/j.issn.1001-7631.2020.01.0060.08对氯甲苯选择性氧化制备对氯苯甲醛魏世明,胡家明,薛艺,张锋南京大学化学化工学院,江苏南京210023摘要:为实现绿色、高效生产对氯苯甲醛,采用氧气氧化对氯甲苯选择性制备对氯苯甲醛,选择以MC(Mid-Century)催化剂为基础催化剂,醋酸作溶剂,筛选助催化剂,探究对氯甲苯转化率和对氯苯甲醛收率的影响因素,并设计正交实验获取优化反应条件。
结果表明反应温度为80 ℃,催化剂用量为对氯甲苯质量的4%,钴盐和锰盐质量比为3:1,对氯甲苯和醋酸体积比为1:2,助催化剂用量为对氯甲苯质量的0.625%时,对氯甲苯转化率可达31.42%,对氯苯甲醛的选择性达81.14%。
在此基础上,通过反应动力学计算,得到了各温度下的反应速率常数和反应活化能。
关键词:对氯甲苯对氯苯甲醛液相氧化动力学中图分类号:O643.32文献标识码:A对氯苯甲醛(PCAD)是一种广泛应用于医药、农药行业的有机中间体[1-2],如何高效、便捷地生产PCAD是化工行业中亟需解决的问题之一。
国内生产PCAD的方法主要是氯化水解法[3-4],该方法对设备要求较高,并且在反应过程中会产生大量的废酸、废水危害环境。
对氯甲苯(PCT)选择性氧化可生产PCAD,其原料易得,原子经济性高[5-6],若是能找到合适的氧化剂及催化剂,可以减少三废的产生甚至不产生三废。
然而PCT的氧化反应是一串联反应,采用MC催化剂(Co/Mn/Br复合催化剂,Mid-Century)和传统工艺条件,PCT的氧化产物主要为氯代苯甲酸(PCA),并不会停留在中间产物PCAD的阶段[7],PCAD在常温下也会自动氧化为PCA,传统的高温氧化工艺会加速该过程,难以达到较高的选择性和收率,同时会产生少量对氯苯甲酸乙酯(PCE)。

甲苯与氯气光照反应方程式
甲苯与氯气的光照反应是一种重要的有机合成反应,在有机化学领域具有广泛的应用。
这种反应是通过光照作用下,甲苯与氯气发生氯代反应,生成对氯甲苯的过程。
其反应方程式如下:
C6H5CH3 + Cl2 → C6H5CH2Cl + HCl
在这个反应中,甲苯(C6H5CH3)与氯气(Cl2)发生光照反应,氯气分子中的氯原子会取代甲苯分子中的氢原子,生成对氯甲苯(C6H5CH2Cl)和氢氯酸(HCl)。
这个反应的机理可以简要解释如下:在光照的作用下,氯气分子中的氯原子会发生光解,生成氯自由基(Cl•)。
氯自由基会与甲苯分子中的氢原子反应,形成氢氯酸和甲苯自由基。
随后,甲苯自由基会与氯自由基发生反应,生成对氯甲苯。
甲苯与氯气的光照反应在有机合成中具有重要意义。
对氯甲苯是一种重要的有机中间体,可以用于制备各种有机化合物,如农药、医药和染料等。
此外,这种反应还可以提供一种有效的方法来合成对氯甲苯,具有一定的工业应用前景。
值得注意的是,这种反应需要在光照条件下进行,光照是促使氯气发生光解的重要条件。
反应过程中还会生成氢氯酸,需要进行适当的处理和中和。
此外,反应中需要控制反应条件,如温度、光照强度和反应时间等,以提高反应的选择性和产率。
总的来说,甲苯与氯气的光照反应是一种重要的有机合成反应,可以合成对氯甲苯这种重要的有机中间体。
通过深入研究这种反应的机理和条件,可以为有机合成化学提供新的方法和思路,推动有机化学领域的发展。


进入20世纪90年代以后,西方发达国家和地区环保压力的不断增加,对有机氯产品发展持谨慎态度,因此生产能力一直没有继续扩大,到了21世纪,氯甲苯下游产品市场的不断扩大,国际市场对氯甲苯衍生出的精细化工中间体需求强劲,刺激和拉动了国内氯甲苯的市场需求。
2000年以来,氯甲苯成为我国众多企业争相发展的有机氯产品,业内很多专家也认为氯甲苯将是未来氯碱企业走精细化道路,建设氯产品精细化工产品树的最具有潜力的基础中间体之一。
从2004年开始,我国掀起了氯甲苯建设的热潮,许多企业新建或扩建生产装置,江苏新业化工有限公司4万t/a氯甲苯装置于2004年投产,江苏丹阳中超化工有限公司和江苏钟腾化工有限公司扩建生产装置均达到2万t/a以上的生产能力,另外,山东,江苏,辽宁等地不少新建装置陆续投产。
氯甲苯通过侧链上氧化,环上氧化,氰化,卤代等反应可以制备许多重要的精细中间体和精细化学品,目前已衍生出数百种农药,医药和染料等产品,以下将应用领域分类介绍一些有发展前景的精细化学品。
1.盐酸噻氯匹定盐酸噻氯匹定(Ticlopidine hydrochloride),化学名称5-(2-氯苄基)-4,5,6,7-四氢噻吩并【3,2-C】吡啶盐酸盐,临床用于与血栓有关的心、脑血管疾病治疗,是由法国Sanofi公司于20世纪70年代研制开发成功,20世纪80年代以来成为欧美、亚洲等的国家治疗心血管疾病的重要药物。
我国也于1989年批准这种药品上市,该药物在治疗缺血性脑血管疾病,外周血管并发缺血性心脏病有独特疗效,同时可以用于治疗多种疾病,不仅疗效显著,且不良反应低,因此被国内心血管专家推荐研制,并列入《国家级化学医药新产品开发指南》中。
我国需求量逐渐上升,噻吩吡啶在栓塞性疾病中治疗地位日趋上升。
国内“八五”和“九五”期间将该药物列为重点开发的药物,但是原料2-噻吩乙胺合成技术难度大,收率较低的问题一直没有得到解决。
其合成方法主要是采用噻吩甲醛为原料,经过噻吩乙胺制得主环4,5,6,7-四氢噻吩并【3,2-C】吡啶,继而与对氯氯苄缩合后成盐。


实验研究对氯苯甲酸的合成张永华(首都师范大学化学系,北京100037) 摘 要:在液相中由氧气氧化对氯甲苯制备对氯苯甲酸,产率92.7%,含量99.2%.研究了催化剂的配比、反应物的浓度、溶剂的含水量以及温度、压力对反应产率的影响.关键词:对氯甲苯;液相氧化;合成;对氯苯甲酸中图分类号:O625.53 文献标识码:A 文章编号:0493-2137(2001)03-0389-03 对氯苯甲酸是用途广泛的有机合成中间体,大量用于有机化工原料、医药和农药的生产[1~4].对氯苯甲酸的制备,一般是以容易得到的对氯甲苯为原料.合成方法有化学试剂氧化法、光氯化水解法、气相氧化法和液相氧气氧化法等.化学试剂氧化法[5~7]、光氯化水解法[8]生产成本高,腐蚀设备严重,产生大量废液、废气,污染环境,逐渐被各工业国淘汰.气相氧化法[9]反应温度高,能耗大,不容易控制,容易产生焦油,收率低.液相氧气氧化法是80年代兴起的方法.它以低级脂肪酸为溶剂,过渡金属化合物和溴化物为催化剂,由空气或氧气氧化烷基芳香烃制取芳香族羧酸,即Amo co法.该法生产成本低,不产生废气、废液,利于环境保护.产品以晶体析出,纯度高,后处理工艺简单,有利于工业化生产[10,11].缺点是低级脂肪酸做溶剂严重腐蚀设备,反应要在较高的压力(2.5~8.0 M Pa)下进行.本文优选了钴盐、锰盐、溴化物构成的催化剂体系,研究了氧化的反应条件,以添加少量丁酸的芳香性卤代烃为溶剂,在低压(0.5~0.7M Pa)下,用氧气氧化对氯甲苯制取对氯苯甲酸,产率92.7%,纯度99.2%,并解决了溶剂腐蚀设备问题.1 实 验1.1 原 料 对氯甲苯、芳香性卤代烃、有机酸为化学纯试剂.乙酸钴、乙酸锰、溴化物为分析纯试剂.1.2 反应式1.3 实验操作 装有电磁搅拌器、热电偶温度控制器和气体导入管的反应釜中加入对氯甲苯18mL(0.15mol),乙酸钴0.58g,乙酸锰0.30g,溴化物0.21g,芳香性卤代烃85m L,丁酸15m L,搅拌,加热,使物料溶解;通入氧气,釜内压力保持0.5~0.7M Pa,125~130℃反应30min,控制温度在115~125℃,反应至釜内压力不再下降为止;冷却,过滤,烘干,得到白色对氯苯甲酸针状晶体22.1g.1.4 产品分析 产品用日本岛津GC-16A气相色谱仪测定其含量为99.2%.经碱溶-盐酸酸化得到白色粉末,用英国8101型数显熔点仪测定m.p为240~241℃.用美国Perkin-Elm er1700型红外光谱仪测定IR,在1400~1600cm-1处有3个强度不等的吸收峰(苯环骨架), 1690cm-1处有尖的强吸收峰(C=O),3000cm-1处有宽的强吸收峰(O-H),750cm-1处有尖的强吸收峰(C=Cl).用日本T X-100型核磁共振仪测定N MR,在 >12.5有(1H)单峰, =7.1~7.3有(2H)双峰, =7.7~7.8有(2H)双峰.确定产品是对氯苯甲酸. 天津大学学报 第34卷 第3期2001年5月JO U RN A L OF T I AN JIN U N IV ERSIT Y V o l.34 No.3 M ay 2001收稿日期:2000-03-17;修回日期:2000-08-15. 作者简介:张永华(1948-),男,副教授.2 合成条件的选择2.1 催化剂的配比 用乙酸钴做主催化剂,乙酸锰做助催化剂,溴化物做引发促进剂,按正交表L 9(33)设计实验,对催化剂体系的配比进行优选.3因素3水平分别是乙酸钴1.0,1.5,2.0mo l;乙酸锰0.4,0.8,1.2mol;溴化物0.5,1.0,1.5m ol .按1.3操作,使反应物浓度为0.5mo l /L(投入对氯甲苯6mL ),保持主催化剂的量是反应物的5%,进行实验.实验条件及结果列于表1.表1 催化剂配比对反应产率的影响Tab .1 Ef fects of ingredient of catalyst on yield实验编号催化剂配比/mol 乙酸钴乙酸锰溴化物产量/g 产率/%1234567891.01.52.01.01.52.01.01.52.00.40.40.40.80.80.81.21.21.21.50.51.01.01.50.50.51.01.56.26.56.36.76.86.44.55.35.178.181.979.384.485.680.656.766.864.2 对表1的数据进行极差分析,表明在实验范围内,催化剂乙酸钴、乙酸锰、溴化物的最佳配比是1.5∶0.8∶1.0(m ol ).进一步实验说明,乙酸钴、乙酸锰、溴化物的配比在1.5∶0.6~0.8∶0.9~1.1(mo l)范围内,合成对氯苯甲酸的产率85.6%~88.2%.2.2 催化剂的用量 为了缩短反应时间,保证反应效果,节省催化剂,按1.3的操作改变催化剂与对氯甲苯的配比,进行了一系列实验,反应所用时间、现象和结果列于表2.表2 催化剂用量对反应产率的影响Tab .2 Ef fect of ratio of catalyst on yield催化剂∶对氯甲苯/%反应需要时间/h反应液颜色产率/%1.52.53.05.07.02.52.02.01.51.4棕黑淡黄淡黄淡黄紫红71.886.996.988.283.1 由表2,催化剂是对氯甲苯的2.5%~5.0%,能获得好的产率,反应时间相差不大,以3%比较合适.少于2.5%时,催化效率不够,反应物焦油化.超过5%时,造成浪费,也使产品进一步氧化分解,降低产率.2.3 反应物的浓度 此反应属于自由基连锁反应.为了使反应顺利进行,有效的措施是稀释反应液,尽量避免自由基之间的碰撞,防止链终止步骤的出现.但是,产物在溶剂中有一定的溶解性.在0~15℃范围内,对氯苯甲酸溶解度是0.8~1.2g.当反应物的浓度低时,溶解在反应液中的产品所占比例高,产率低.为了获得好的产率,按1.3的操作,改变对氯甲苯在反应液中的浓度,进行一系列实验,其结果列于表3.表3 反应物浓度对反应产率的影响Tab .3 Ef fect of concentration of reaction substrat on yield反应物浓度/(mol/L )投料量/g产品产量/g反应产率/%0.511.01.52.03.06.412.819.325.237.87.014.422.029.040.488.290.792.392.786.0 由表3看出对氯甲苯的浓度在1.5~2.0m ol /L 时产率最高.气相色谱跟踪测定表明,对氯甲苯的浓度高于3.0m ol/L 时,氧化不完全,产率下降.2.4 溶剂的含水量 对Amo co 法的研究,有文献报道水对反应产率有明显的影响,建议加入脱水剂来提高产率.作者按反应物的不同浓度往溶剂中加入不同量的水,按1.3操作,进行了一系列实验,其结果列于表4.表4 溶剂含水量对反应产率的影响Tab .4 Eff ect of water content in solvent on yield反应物浓度/(mol ・L -1)0.5 1.02.0溶剂加水量/%00.010.10.82.500.800.8反应产率/%86.987.488.288.283.190.792.692.792.7 由表4看出,溶剂含少量水,反应产率有所提高.含水量高到一定程度,产率下降.笔者认为,形式上Amo co 法是气、液相氧化的多相反应,实际上是均相反应.催化剂在溶剂中溶解形成溶液,氧气在一定压力下借助与脂肪酸间的氢键作用溶于溶剂,催化氧化反应在均一的液相中进行.反应中,溴化物是自由基引发促进剂,它以下列方式加速自由基的产生: Br -作为配位基与Co 2+结合生成Co 2+Br -,O 2与・390・天津大学学报 2001年 第34卷 第3期 Co2+Br-络合后,配位基Br-通过中心金属钴将电子转移给O2,形成超氧基团和自由基Br.自由基Br从烃分子中夺取氢,引发出径自由基R.溶剂中含有少量水,能够促进溴化物解离出Br-离子,加速自由基的形成.在反应液中加入少量乙酸酐,以脱去催化剂所带结晶水和溶剂中的微量水分.发现反应在140℃才能引发,产率只有78%.溶剂中的水分超过它在溶剂中的溶解度时,体系形成多相,催化剂转入水相,催化效率下降.反应物的浓度对反应产率的影响可能与反应生成的水有关.反应物浓度低,氧化生成的水在反应温度下可以溶于溶剂中.反应物浓度高,氧化生成的水超过溶剂的溶解能力,液体分层,催化剂进入水层,失去催化能力.催化剂所含结晶水和工业级溶剂含水分足以促进溴化物解离,使氧化反应顺利进行.2.5 反应的温度和压力 液相催化氧气氧化芳香烃,一般用加压鼓泡法.它要求反应液有一定的深度,同时需要氧气回收和加压装置.本合成法采用密闭容器加压氧化,工作压力0.5~0.7MPa,足以保证氧气在溶液中的溶解度和氧化效率.反应温度控制在115~125℃为宜.温度低,反应速度慢;温度高,焦油化严重.值得注意的是,反应中温度不可忽高忽低,防止自由基瘁灭.3 结 论 由乙酸钴、乙酸锰、溴化物催化,氧气氧化对氯甲苯,制取对氯苯甲酸可以在添加丁酸的芳香性卤代烃中进行.反应温度115~125℃,压力0.5~0.7MPa.丁酸在反应液中≤20%时,对不锈钢反应釜没有明显的腐蚀作用.反应液经活性白土脱色或蒸馏可以重复使用.用此法生产对氯苯甲酸设备与操作简单,无三废排放,能耗少,成本低,便于工业化生产.参考文献:[1] 刘方明,于建新,刘育亭,等.3-(2′-苯基-1′,2′,3′-连三唑-4′-基)-6-烷基芳基均三唑并[3,4-b]-1,3,4-噻二唑的合成[J].化学学报,1998,56(6):618-624.[2] O r Y S,Phen L T,Chu D T et al.Pr epa ration of tr icyclicer yt hr om ycins as Bacter icides[P].W O9830574,1998. [3] 孙昌俊,王义贵,李洪祥,等.糖苷合成研究(ⅤⅡ)1-0- -D芳酰基-2,3,4-三-0-乙酰基葡萄糖醛酸甲酯的合成及其生物学活性[J].山东大学学报,1996,31(3):327-331.[4]Shrivastav a A K,K umar S,Sabkar P C.2-amino-4-ary lth-iazoles and their thiazo ly la mides as antifung al agents[J].J Inst Chem(India),1997,69(4):113-115.[5] Sasson Y,Zappi G D,N eumann R.L iquid-phase ox idationo f deactiv ated methy lbenzenes by aqueo us sodiumhy po chlor it e catalyzed by r uthenium salts under phase t ransfer cataly tic co nditions[J].J O rg Chem,1986,51(15):2880-2883.[6] Jur sic B.Sur fact ant assist ed permang anate ox idatio n o faro matic co mpo unds[J].Can J Chem,1989,67(9):1381-1383.[7] 王 旗.芳香族羧酸的制备[P].CN1104626,1995.[8] K aminski J,Chmielo w iec U,Duczma le W,et al.M ethodfo r manufacturing pur e o-and p-chlo ro benzoic acids[P].P L155519,1992.[9] A nto l M,Cv engr osov a Z,V r abel I,et al.O xidat ion o falky lar om atic hydro carbons ov er V2O5-Sb2O3/T iO2cat a-ly st[J].Collect Czech Chem Co mmun,1997,62(9):1481-1490.[10]F eld M ar cel.Halobenzo ic acids fr om ring halo genatedt oluene[P].DE3308448,1984.[11]R oehrscheid F,Gr oetsch G.Pr o cess for prepar ing br omi-nated o r chlo rinated ar omat ic car bo x ylic acids[P].EP 713856,1996.SYNTHESIS OF p-CHLOROBENZOIC ACIDZHANG Yong-hua(Dept.of Chemistr y,Capital No rma l U niv ersity,Beijing100037,China)Abstract:p-chlo ro benzoic acid w as pr epar ed by o x ida tio n o f p-chlor ot oluene w ith diox y gen in liquid-phase.T he y ield can reach92.7%,pur it y99.2%.T he effects of the ing redients o f the cata ly st,co ncentr atio n of the r eactio n subst rat,w ater co ntent in the solvent and t emper atur e,pressure o n t he y ield ar e studied.Keywords:p-chlo ro toluene;liquid-phase ox idation;synthesis;p-chlor obenzo ic acid ・391・ 天津大学学报 张永华:对氯苯甲酸的合成。

![甲苯氯化制备对氯甲苯和邻氯甲苯的方法[发明专利]](https://uimg.taocdn.com/54f25eba690203d8ce2f0066f5335a8102d26619.webp)
[19]中华人民共和国国家知识产权局[12]发明专利申请公布说明书[11]公开号CN 101497552A [43]公开日2009年8月5日[21]申请号200910030310.7[22]申请日2009.03.19[21]申请号200910030310.7[71]申请人江苏钟腾化工有限公司地址212300江苏省丹阳市化工路3号[72]发明人钟华 陆敏山 刘巧宝 孙建平 潘晓鑫 [74]专利代理机构镇江京科专利商标代理有限公司代理人夏哲华[51]Int.CI.C07C 25/02 (2006.01)C07C 17/12 (2006.01)C07C 17/383 (2006.01)权利要求书 3 页 说明书 8 页 附图 3 页[54]发明名称甲苯氯化制备对氯甲苯和邻氯甲苯的方法[57]摘要甲苯氯化制备对氯甲苯和邻氯甲苯的方法,是原料甲苯进干燥器干燥后和氯气在催化剂的作用下,进行直接氯化,得到混氯甲苯、副产盐酸和少量残渣,氯化工艺得到的混氯甲苯作为原料送到混氯甲苯分离工艺,经精密分馏得到邻氯甲苯,对氯甲苯和少量的残液。
采用本发明,塔式固定床循环氯化工艺,这保证了气液接触更加充分,反应热能及时移出,反应效果更佳,副产物减少,解决了传统吸收过程中正压或常压系统中氯化氢气体泄漏导致的环境污染问题。
避免了废水的产生,两塔串联连接的精馏分离方式,降低了塔的安装高度,提高了分离效率,最终产品对氯甲苯和邻氯甲苯的纯度均≥99.9%wt。
200910030310.7权 利 要 求 书第1/3页 1、甲苯氯化制备对氯甲苯和邻氯甲苯的方法,是原料甲苯进干燥器干燥后与氯气在催化剂的作用下,进行直接氯化得到混氯甲苯、副产盐酸和少量残渣,氯化工艺得到的混氯甲苯作为二次原料送到混氯甲苯分离工艺,经精密分馏得到邻氯甲苯、对氯甲苯和少量的残液;其特征在于:1)、所述的直接氯化法:是以甲苯为原料,经干燥器(1)干燥后与辅助催化剂在配料槽(2)内搅拌均匀,将物料打入氯化塔(3)内,启动氯化循环泵,缓慢开启氯气进料流量计,在催化剂的作用下进行氯化反应,根据反应塔内温度控制氯气流量,塔温控制在25℃-72℃之间,绝压压力为0.06~0.095Mpa,氯化液循环氯化,通过冷却器(4)移出反应热;氯化产生的氯化氢气体通过尾气吸收装置生成盐酸,在取样检测达到指标值后停止氯化,向蒸馏釜(5)出料,蒸馏釜(5)釜温60℃-155℃,釜内绝压0.05~0.085Mpa,氯化液在蒸馏釜(5)内先利用真空抽吸和蒸汽加热进行曝气,脱除溶解的氯化氢气体,然后再经过蒸馏去除催化剂,进入脱甲苯塔(6),脱除未反应的甲苯和轻组分后得到主要含对氯甲苯和邻氯甲苯的混合物即混氯甲苯;2)、所述的混氯甲苯分离工艺:是将混氯甲苯连续进入塔内温度为70℃-130℃,绝压压力为0.003~0.025Mpa的初分塔(10)顶部,塔顶富含邻氯甲苯的混合物进入邻塔I(12),邻塔I(12)的汽相直接通入邻塔I I(14)底部,邻塔I(12)和邻塔I I(14)的塔内温度为70℃-125℃,绝压压力为0.008~0.028Mpa,邻塔II(14)底部物料通过循环泵泵入邻塔I(12)顶部,邻塔I I(14)顶部得到产品邻氯甲苯其含量≥99.9%wt,邻塔I(12)塔底馏分进初分塔(10)循环分离;同时初分塔(10)底部富含对氯甲苯的物料进入对塔II(17)顶部,对塔II塔(17)顶得到轻组分进初分塔循环分离,对塔II(17)塔底物料通过循环泵泵入对塔I(15)顶部,对塔I(15)和对塔I I (17)的塔内温度为80℃-135℃,绝压压力为0.005~0.020Mpa,对塔I(15)的汽相直接通入对塔I I(17)的底部,对塔I(15)底部得到对氯甲苯中间料进入贮槽(18),对氯甲苯中间料泵入塔内温度为90℃-140℃,绝压压力为0.003~0.015Mpa的对精塔(19)顶部连续精馏,塔顶得到含量≥99.9%wt高纯度的对氯甲苯,塔底为残液。
![一种由对氯甲苯合成对氯苯甲醛的方法[发明专利]](https://uimg.taocdn.com/63a29e06effdc8d376eeaeaad1f34693daef10ea.webp)
(10)申请公布号(43)申请公布日 (21)申请号 201410811658.0(22)申请日 2014.12.24C07C 47/55(2006.01)C07C 45/43(2006.01)C07C 45/42(2006.01)(71)申请人常熟市新华化工有限公司地址215500 江苏省苏州市常熟市海虞镇福山北8号(72)发明人窦建华 陆志忠 徐俊德(54)发明名称一种由对氯甲苯合成对氯苯甲醛的方法(57)摘要本发明公开了一种由对氯甲苯合成对氯苯甲醛的方法,将对氯甲苯在引发剂存在下160~180℃和氯气反应3~8h ,生成对氯苄基氯,然后催化水解6~12h ,即得对氯苯甲醛;本发明的由对氯甲苯合成对氯苯甲醛的方法,原料廉价,操作简便。
(51)Int.Cl.(19)中华人民共和国国家知识产权局(12)发明专利申请权利要求书1页 说明书2页(10)申请公布号CN 104447250 A (43)申请公布日2015.03.25C N 104447250A1.一种由对氯甲苯合成对氯苯甲醛的方法,其特征在于,包括下述步骤:将对氯甲苯在引发剂存在下160~180℃和氯气反应3~8h,生成对氯苄基氯,然后催化水解6~12h,即得对氯苯甲醛。
2.根据权利要求1所述的由对氯甲苯合成对氯苯甲醛的方法,其特征在于:所述的引发剂用量相对于对氯甲苯为1~1.5wt%。
3.根据权利要求1所述的由对氯甲苯合成对氯苯甲醛的方法,其特征在于:所述的催化剂用量相对于对氯苄基氯为0.5~0.8wt%。
4.根据权利要求1所述的由对氯甲苯合成对氯苯甲醛的方法,其特征在于:将对氯甲苯在引发剂存在下170℃和氯气反应6h。
5.根据权利要求1所述的由对氯甲苯合成对氯苯甲醛的方法,其特征在于:生成对氯苄基氯后,催化水解8h。
一种由对氯甲苯合成对氯苯甲醛的方法技术领域[0001] 本发明属于有机合成技术领域,具体涉及一种由对氯甲苯合成对氯苯甲醛的方法。

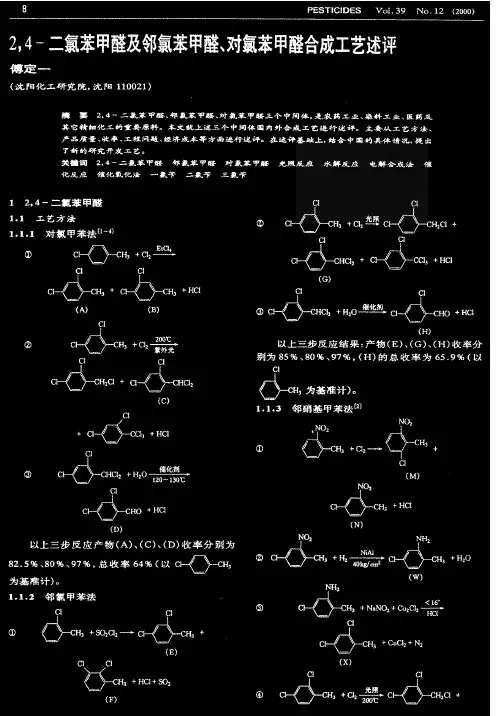
对氯甲苯合成的实验研究对氯甲苯是农药、医药、染料和其它精细有机化工产品的重要原料和中间体。
通过对氯甲苯侧链上的氯化、氧化、氨氧化和环上的氯化、硝化、磺化、Friedel-Crafts反应、氯甲基化等反应,可以衍生出一系列的重要精细化工中间体,通过这些中间体可以开发出100多种农药、医药、染料产品。
如农药杀草丹、多效唑、氟乐灵、拟除草菊酯类杀虫剂;医药消炎痛;染水染染色基等。
对氯甲苯传统的生产方法是以对甲苯胺为原料,经重氮化生成重氮盐,然后在氯化亚铜催化剂存在下,重氮盐与浓盐酸反应生成对氯甲苯。
该方法生产成本高,废量大,设备腐蚀和污染严重。
目前国内外的生产厂多采用氯气与甲苯反应生产邻氯甲苯与对氯甲苯的混合物(简称混氯),然后再通过精馏的方法,将邻氯甲苯与对氯甲苯分离。
现采用天然活性沸石作为甲苯氯化催化剂,加入助催化剂提高其催化活性、耐水性和使用寿命,并使邻对比大幅降低,间位异构体减少到允许水平。
笔者探索了化过程各因素对反应的影响。
一、实验方法1、仪器与试剂。
实验采用的主催化剂为天然活性沸石,为一种浅灰或浅棕色粉状粘土,主要成分是Si02、A1203、Fe203Mg0、K20,处理后供使用;助催化剂A,铁盐,试剂级;助催化剂B,卤代乙醇,试剂级;助催化剂C,硫化物,工业级;助催化剂D,硫的氯化物,工业级;甲苯,石油一级;氯气,工业级,纯度>9915%。
反应后滤液进行谱分析,102G气相色谱仪,热导池检测,N2为载气,流速18mL/min,柱温90℃,气化室温度210℃。
黑色250mL三口烧瓶;磁力搅拌器;冷凝管;恒温水浴锅。
1.12实验方法实验流程如图1所示。
氯化反应器为250mL三口烧瓶,三口分别插温度计、尾气冷凝管和通氯管。
实验时将100mL甲苯和催化剂加入釜内,氯气经毛细管流量计计量后从釜底均匀通入,磁力搅拌器搅拌,恒温水浴控温,尾气经冷凝回收甲苯并经碱液吸收后从水冲泵抽走。
用淀粉碘化钾试纸检验、控制尾气中不含游离氯。
对氯甲苯的合成
一、实验目的
1.通过本实验学习对氯苯胺的制备原理和方法以及重氮化反应操作。
2.进一步熟练掌握水蒸汽蒸馏的安装和操作。
3.熟练掌握用冰水控温法。
二、实验原理
对氯甲苯是一种有机合成原料,外观为无色透明液体,有特殊气味,能于醇、醚、苯等,微溶于水。
比重:1. 0697,熔点:7. 50C,沸点162℃。
用于医药、农药、染料方面。
可生产对氯氯苄、对氯苯甲醇、对氯氰节、对氯苯甲醛、对二氯甲酸、2, 4-二氯甲苯、2, 4一二氯苯甲醛、氰戊菊酷、杀菌剂等。
工业上有两种生产方法:一是甲苯经硝化、还原、重氮化制备;二是甲苯经氯化制备。
本品有毒,对呼吸道有损伤,对眼、鼻有刺激作用,避免用手直接接触,非密闭场所要穿戴防护用品。
本实验采用以对甲苯胺为原料,经重氮化合成对氯甲苯的方法。
三、试剂与仪器
试剂:对甲苯胺、五水硫酸铜、氯化钠、氢氧化钠、盐酸、亚硫酸氢钠和硝酸钠都是化学纯。
仪器:标准磨口玻璃仪器、电动搅拌器、可调功率电炉。
四、实验步骤
1.氯化亚铜溶液的制备
在400m1烧杯内,将27. 5g五水硫酸铜和10. 0g氯化钠溶解于100m1水中,加热到60一70℃,减压过滤,除去不溶的杂质,得溶液A.将6. 5g亚硫酸氢钠和3. 0g氢氧化钠溶解于50水中,也加热到60-70℃,减压过滤,除去不溶的杂质,得溶液B。
在搅拌下,缓慢地把溶液B加到溶液A中,析出白色的氯化亚铜。
冷却到室温,用含少量的亚硫酸氢钠的水以倾泻法洗涤氛化亚铜,然后将己
冷却到水温2℃以下的30m1浓盐酸倒入氯化亚铜中。
2.重氮盐的制备
在250m1三口圆底烧瓶中,依次放入10. 7g对甲苯胺、lOml水和40m1浓盐酸,搅拌,加热到60℃使对甲苯胺完全溶解,再用冰盐浴冷却到5℃以下。
在50m1烧杯中,把7. 0g亚硝酸钠溶解于20m1水中,冷却到5℃以下后,把它放入50m1分液漏斗中.在搅拌下,把亚硝酸钠溶液慢慢地滴入三口圆底烧瓶中,保持反应温度不超过5℃,近终点时,重氮化反应速度较慢。
亚硝酸钠溶液滴加速度控制在每分钟1一2滴,不时用碘化钾淀粉试纸来检验终点。
如果试纸立刻变蓝色,就表示重氮化反应已完成。
3.对氯甲苯的制备
将重氮盐溶液缓缓倒入己冷却至0℃的氛化亚铜盐酸溶液中,并搅拌,用冰水浴冷却,使反应温度控制在15℃以下。
这时,有深红色悬浮物析出。
大约10分钟后,撤去冰水浴,在室温下反应2. 5小时,再用水浴慢慢加热到60℃,保持半小时,直到没有气泡放出为止.将反应混合物进行水蒸气蒸馏,直到馏出液中没有油珠时为止。
把馏出液倒入分液漏斗中,分离出粗对级甲苯。
用适量的用水洗涤一次,分离出对抓甲苯,用无水氛化钙干燥后,进行蒸馏,收集160一164℃的馏分。
得有特殊气味、无色透明液体,产量为10. 8g,含量为97. 2%。
五、注意事项
1.配置各溶液时各量一定要标准。
2.加入亚硫酸氢钠溶液时一定要振摇,否则形成的褐色沉淀易结块,影响氯化亚铜的质量。
3.制备氯化亚铜时静置时白色的氯化亚铜沉淀完全,倾倒上层液体时要小心不要将沉淀倒出。
4.氯化亚铜易被氧化成有色的二价铜盐,制备好以后应密闭冷却保存。
5.制备重氮盐时一定要保持好温度。
在加入90-95%的亚硝酸钠溶液后即可用试纸检验,变蓝则不再继续加入。
6.分解重氮盐-CuCl复合物宜室温多放置,加热分解。
六、思考题
1.重氮化反应在有机合成中的用途。
2.在分离纯化过程中,碱洗、酸洗的目的分别是为了除去什么?3.抽滤时怎样洗涤产品?
4.为什么不直接将甲苯氯化而是通过本实验的方法来制备对氯甲苯。