内科学(第七版)造血系统疾病第十六 章凝血障碍性疾病
- 格式:doc
- 大小:558.00 KB
- 文档页数:12
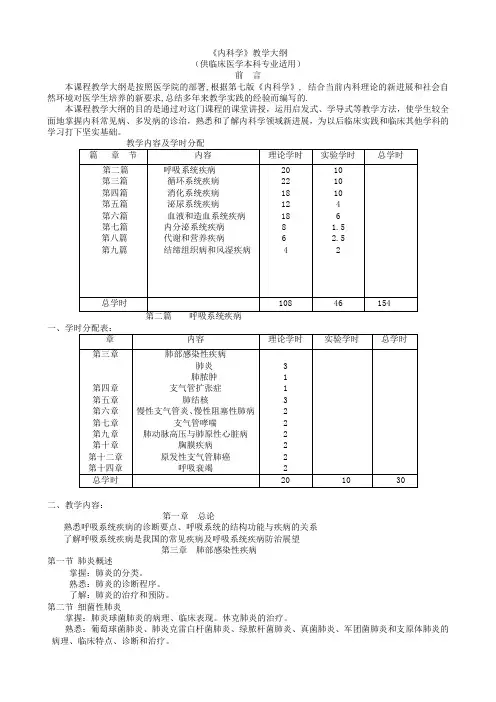
《内科学》教学大纲(供临床医学本科专业适用)前言本课程教学大纲是按照医学院的部署,根据第七版《内科学》, 结合当前内科理论的新进展和社会自然环境对医学生培养的新要求,总结多年来教学实践的经验而编写的.本课程教学大纲的目的是通过对这门课程的课堂讲授,运用启发式、学导式等教学方法,使学生较全面地掌握内科常见病、多发病的诊治,熟悉和了解内科学领域新进展,为以后临床实践和临床其他学科的学习打下坚实基础。
二、教学内容:第一章总论熟悉呼吸系统疾病的诊断要点、呼吸系统的结构功能与疾病的关系了解呼吸系统疾病是我国的常见疾病及呼吸系统疾病防治展望第三章肺部感染性疾病第一节肺炎概述掌握:肺炎的分类。
熟悉:肺炎的诊断程序。
了解:肺炎的治疗和预防。
第二节细菌性肺炎掌握:肺炎球菌肺炎的病理、临床表现。
休克肺炎的治疗。
熟悉:葡萄球菌肺炎、肺炎克雷白杆菌肺炎、绿脓杆菌肺炎、真菌肺炎、军团菌肺炎和支原体肺炎的病理、临床特点、诊断和治疗。
了解:院内外感染所致细菌性肺炎在菌种上的不同。
第三节支原体肺炎(自学)熟悉:支原体肺炎的病理、临床特点、诊断和治疗。
第四节肺脓肿掌握:肺脓肿的诊断、治疗原则和方法。
熟悉:肺脓肿的病因、发病原理和鉴别诊断。
了解:外科治疗的适应症。
第四章支气管扩张症掌握:支气管扩张症的诊断、鉴别诊断、治疗及咯血的处理熟悉:支气管扩张症的病因及发病机理了解:外科治疗的适应症。
第五章肺结核掌握:结核的发生与发展过程与变态反应和免疫力的关系、诊断及鉴别诊断要点,抗结核药物的正确使用、大咯血的处理。
熟悉:肺结核X线特点并与病理变化的关系、痰结核菌的检查方法、原则和方法、结核菌素试验、卡介苗接种。
了解;抗结核结核病控制策与措施。
第六章慢性支气管炎、慢性阻塞性肺疾病掌握:慢性支气管炎、慢性阻塞性肺疾病的概念、诊断好的鉴别诊断、治疗,特别是慢性阻塞性肺疾病的肺功能的检查特点、严重程度分级以及并发症。
熟悉:;慢性阻塞性肺疾病的发生和发展规律,特别是由慢性支气管炎发展成为肺气肿的过程及其病理变化。


第七版内科护理学简答题主观题背诵第五章血液系统疾病病人的护理1.简述血液病的临床类型,并举例说明(1)RBC疾病:如缺铁性贫血。
(2)粒细胞疾病:如WBC 减少与粒细胞缺乏。
(3)单核细胞和巨噬细胞疾病:如单核细胞增多症。
(4)淋巴细胞和浆细胞疾病:如各类淋巴瘤。
(5)造血干细胞疾病:如再生障碍性贫血。
(6)脾功能亢进。
(7)出血性及血栓性疾病:如ITP。
2.红细胞、白细胞、血小板计数和血红蛋白测定成人红细胞计数,男性为(4.0~5.5)X1012/L,女性为(3.5~5.0)×1012/L:血红蛋白男性为120~160g/L,女性为110~150g/L。
正常成人白细胞计数为(4~10)×109/L,血小板正常值(100~300)×10L3.皮肤出血的预防与护理保持床单平整,衣者轻软、宽松:避免肢体的碰撞或外伤。
沐浴或清洗时,避免水温过高和过于用力擦洗皮肤:勤剪指甲,以免抓伤皮肤。
高热病人禁用酒精(温水)拭浴降温。
各项护理操作动作轻柔:尽可能减少注射次数:静脉穿刺时,应避免用力拍打及揉擦局部,结扎压脉带不宜过紧和时间过长:注射或穿刺部位拔针后需适当延长按压时间,必要时局部加压包扎。
此外,注射或穿刺部位应交替使用,以防局部血肿形成。
4.鼻出血的预防与护理(1)防止鼻黏膜干燥而出血:保持室内相对湿度在50%~60%左右。
(2)避免人为诱发出血:指导病人勿用力擤鼻,以防止鼻腔内压力增大而导致毛细血管破袋出血或渗血:避免用手抠鼻痴和外力撞击鼻部。
(3)少量出血时,可用棉球或明胶海绵填塞,无效者可用0.1%肾上腺素棉球或凝血酶棉球填塞,并局部冷敷。
出血严重时,尤其是后鼻腔出血,可用凡士林油纱条行后鼻腔填塞术,术后定时用无菌液状石蜡滴入。
5.口腔、牙龈出血的预防与护理指导病人用软毛牙刷刷牙,忌用牙签剔牙:尽量避免食用煎炸、带刺或含坚硬骨头的食物、带硬壳的坚果类食品以及质硬的水果(如甘蔗)等;进食时要细嚼慢咽,避免口腔黏膜的损伤。


贫血概述一、定义贫血( Anemia )是指人体外周血红细胞容量减少,低于正常范围下限的一种常见临床症状。
为测量方便,临床常以血红蛋白( Hemoglobin,Hb )浓度来代替。
外周血中单位容积内Hb浓度、红细胞计数( RBC )和 / 或血细胞比容( HCT )低于相同年龄、性别和地区的正常标准,可诊断贫血。
二、诊断标准1. 我国诊断贫血标准:海平面地区男: Hb < 120g/L RBC < 4.5 × 1012/L 和 / 或 HCT < 0.42 ,女: Hb < 110g/L RBC < 4.0 × 1012/L 和/ 或 HCT < 0.37孕妇: Hb < 100g/L RBC < 3.5× 1012/L 和/ 或 HCT < 0.302. 国外诊断贫血标准:在海平面地区, Hb低于下述水平诊断贫血:6 个月~ <6 岁: Hb<110g/L 6~14 岁: Hb<120g/L成年男性: Hb<130g/L 成年女性: Hb<120g/L孕妇: Hb<110g/L3. 婴儿、儿童、妊娠妇女的 Hb 浓度较成人低。
多与造血营养物质的需求增多,摄入相对不足有关。
4. 久居高原地区的居民平均 Hb 浓度较海平面居民高。
与高原地区缺氧状态刺激促红素生成增多有关。
5. 某些状况下,血液稀释(血浆容量增多,如充血性心衰、巨球蛋白血症、妊娠等;补液过多等)可表现 Hb 浓度相对下降;血液浓缩(失血、脱水等),则可表现血红蛋白浓度相对增高。
所以,需结合多方面因素综合分析,方可正确作出贫血的诊断。
6. 老年人诊断贫血的指标可适度降低。
三、贫血的临床分类1. 按贫血进展速度:急性贫血( acute anemia ):常见于失血性贫血;慢性贫血( chronic anemia ):缺铁性贫血、巨幼细胞性贫血多表现为慢性贫血。
2. 按血红蛋白浓度分:轻度贫血 90g/L < Hb< 正常低限;中度贫血 60g/L<Hb<90g/L;重度贫血 30g/L<Hb<60g/L;极重度贫血 Hb<30g/L。






【内科学血液系统】凝血障碍性疾病考点总结●血友病●病因与遗传规律●病因●血友病 A 又称FⅧ缺乏症,是临床上最常见的遗传性出血性疾病,FⅧ基因位于 X 染色体长臂末端●FⅧ——抗血友病球蛋白;血友病A又称抗血友病球蛋白缺乏症●血友病 B 又称遗传性FⅨ缺乏症,FⅨ基因位于 X 染色体长臂末端●遗传规律●血友病 A、B 均属 X 连锁隐性遗传性疾病●临床表现●出血●血友病 A 出血较重,血友病 B 则较轻●血友病 A 分为3 型●重型:FⅧ:C 活性低于 1%●中型:FⅧ:C 活性 1% ~5%●轻型:FⅧ:C 活性 6% ~ 30%●特点●与生俱来,伴随终身●常表现为软组织或深部肌肉内血肿;●负重关节如膝、踝关节等反复出血甚为突出,最终可致关节肿胀、僵硬、畸形●血肿压迫症状及体征●实验室检查●筛选试验●出血时间、凝血酶原时间(PT)12s~14s、血小板计数及血小板聚集功能正常, APTT 延长23s~37s●确诊试验●FⅧ活性测定辅以FⅧ:Ag 测定●FⅨ活性测定辅以FⅨ:Ag 测定●vWF:Ag 测定(血友病病人正常),可与血管性血友病鉴别●基因诊断试验●诊断与鉴别诊断●诊断参考标准●血友病 A●临床表现●男性病人,有或无家族史,有家族史者符合 X 连锁隐性遗传规律●关节、肌肉、深部组织出血,可呈自发性●实验室检查●出血时间、血小板计数及 PT 正常●APTT 延长,能被钡吸附正常血浆纠正●钡吸附正常血浆中含有FⅧ,正常血清含有FlX,血友病A缺乏FⅧ,故可被钡吸附正常血浆纠正而血友病B不能,血友病B能被正常血清纠正●FⅧ: C 水平明显低下●vWF:Ag 正常●血友病 B●临床表现●基本同血友病人但程度较轻●实验室检查●出血时间、血小板计数及 PT 正常●APTT重型延长,轻型可正常●FlX抗原及活性减低或缺乏●鉴别诊断●治疗与预防●原则●加强自我保护,预防损伤出血极为重要●尽早有效地处理病人出血,避免并发症的发生和发展●禁用阿司匹林、非甾体类抗炎药及其他可能干扰血小板聚集的药物●家庭治疗及综合性血友病诊治中心的定期随访●出血严重病人提倡预防治疗●一般疗法●替代疗法●补充缺失的凝血因子,它是防治血友病出血最重要的措施●主要制剂有基因重组的纯化FⅧ、FⅧ浓缩制剂、新鲜冷冻血浆、冷沉淀物(FⅧ浓度较血浆高5 ~ 10 倍)以及凝血酶原复合物●最低止血要求FⅧ: C 或 FIX水平达 20% 以上,出血严重或欲行中型以上手术者,应使FⅧ或 FIX活性水平达 40% 以上●FⅧ剂量(U)=体重( kg)×所需提高的活性水平(% ) ÷2●FIX剂量(U)=体重(kg) x所需提高的活性水平(%)●其他药物治疗●去氨加压素●可促进内皮细胞释放储存的 vWF 和FⅧ●抗纤溶药物●氨基己酸和氨甲环酸●家庭治疗●外科治疗●基因疗法●预防●血管性血友病●显性遗传,以自幼发生的出血倾向、出血时间延长、血小板黏附性降低、瑞斯托霉素诱导的血小板聚集缺陷,及血浆vWF抗原缺乏或结构异常为特点●病因和发病机制●vWF●与FⅧ:C 以非共价键结合成 vWF-FⅧ:C 复合物,vWF 增加FⅧ:C稳定性、防止其降解,并促进其生成及释放●vWF 在血小板与血管壁的结合中起着重要的桥梁作用●vWF基因位于 12 号染色体短臂末端●获得性血管性血友病●最常见的是产生具有抗 vWF 活性的抑制物,主要为 IgG;其次为肿瘤细胞吸附 vWF,使血浆 vWF减少;另外,抑制物可与 vWF 的非活性部位结合形成复合物,加速其在单核-巨噬细胞系统的破坏●临床表现●出血以皮肤黏膜为主,如鼻出血、牙龈出血、瘀斑等,外伤或小手术(如拔牙)后的出血也较常见●男女均可发病,女性青春期病人可有月经过多及分娩后大出血●出血可随年龄增长而减轻,此可能与随着年龄增长而 vWF 活性增高有关●自发性关节、肌肉出血相对少见●实验室检查●出血筛选检查●血小板计数正常,出血时间延长或阿司匹林耐量试验延长●诊断试验●血浆 vWF 抗原测定( vWF : Ag),血浆 vWF 瑞斯托霉素辅因子活性( vWF : RCo)以及血浆FⅧ凝血活性(FⅧ:C)测定●血管性血友病多数vWF抗原降低,而Ⅷ:C仅轻度降低,故Ⅷ:C/ⅧR:A比例应升高●vWD 分型诊断试验●血浆 vWF 多聚体分析●瑞斯托霉素诱导的血小板聚集 (RIPA)●血浆 vWF 胶原结合试验( vWF: CB)●④血浆 vWF 因子回结合活性(vWF:FⅧB)●诊断与分型●诊断要点●有或无家族史●有自发性出血或外伤、手术后出血增多史●血浆 vWF:Ag<30% 和(或)vWF:RCo<30% ;FⅧ:C<30% 见于 2N 型和 3型 vWD●排除血友病、获得性 vWD、血小板型 vWD、遗传性血小板病等●鉴别诊断●分型●治疗●去氨加压素( DDAVP)●对 3 型 vWD 无效;对 2B 型 vWD 慎用●替代治疗●选用血源性含 vWF 浓缩制剂或重组 vWF 制剂●其他治疗●抗纤溶药物。
---------------------------------------------------------------最新资料推荐------------------------------------------------------内科学(第七版)造血系统疾病第十六章凝血障碍性疾病内科学(第七版)造血系统疾病第十六章凝血障碍性疾病第十六章凝血障碍性疾病凝血障碍性疾病是凝血因子缺乏或功能异常所致的出血性疾病。
凝血障碍性疾病大致可分为先天性和获得性两类。
前者与生俱来,多为单一性凝血因子缺损,如血友病等;后者发病于出生后,常存在明显的基础疾病,多为复合性凝血因子减少,如肝病性出血等。
第一节血友病血友病(hemophilia)是一组因遗传性凝血活酶生成障碍引起的出血性疾病,包括血友病 A、血友病 B 及遗传性FⅪ缺乏症,其中以血友病 A 最为常见。
血友病以阳性家族史、幼年发病、自发或轻度外伤后出血不止、血肿形成及关节出血为特征。
血友病的社会人群发病率为 5~10/10 万,婴儿发生率约1/5000。
血友病A、B及遗传性FⅪ缺乏的比较发病率为16:3:1,我国的血友病中,血友病 A 约占 80%,血友病 B 约占 15%,遗传性FⅪ缺乏症则极少见。
【病因与遗传规律】 (一)病因血友病 A 又称遗1 / 15传性抗血友病球蛋白缺乏症或FⅧ:C 缺乏症。
FⅧ由两部分组成:即。
FWⅢ凝血活性部分(FⅧ:C)和 vWI)因子(vWF)。
两者以复合物形式存在于血浆中。
前者被激活后参与 FX 的内源性激活;后者作为一种黏附分子参与血小板与受损血管内皮的黏附,并有稳定及保护FVⅢ:C 的作用。
FⅧ:C 基因位于 X 染色体长臂末端(Xq28),当其因遗传或突变而出现缺陷时,人体不能合成足量的FⅧ:C,导致内源性途径凝血障碍及出血倾向的发生。
血友病 B 又称遗传性FⅨ缺乏症。
FⅨ为一种单链糖蛋白,被Ⅺa 等激活后参与内源性 FX 的激活。
凝血功能障碍凝血功能障碍性疾病(coagulation diaorders)是指凝血因子缺乏或功能异常所致的出血性疾病。
可分为遗传性和获得性两大类。
遗传性凝血功能障碍一般是单一凝血因子缺乏,多在婴幼儿期即有出血症状,常有家族史。
获得性凝血功能障碍较为常见,患者往往有多种凝血因子缺乏,多发生在成年,临床上除出血外尚伴有原发病的症状及体征。
一、血友病血友病(hemophilia)是最常见的一组遗传性凝血因子缺乏症,可分为血友病甲(因子Ⅷ促凝成分即Ⅷ缺乏)及血友病乙(因子Ⅸ缺乏)两型。
因子Ⅷ:C及因子Ⅸ的生物合成基因均位于X染色体,故称X链疾病,两者均为X染色体伴性隐性遗传,男性发病,女性传递。
女性携带者虽有不同程度的因子Ⅷ:C或因子Ⅸ活性减低,但一般无出血症状。
约1/3患者查无家族史,可能是家族中男性少或隔代遗传而被忽视,也可能是基因突变所致。
(一)临床表现主要表现为出血,以软组织、肌肉、负重关节出血为特征。
通常自幼儿期即有出血倾向,轻型可在青少年甚至成年才被诊断。
出血症状出现越早,病情越重。
患者可表现为轻微外伤或手术后严重出血,往往在拔牙或小手术时出血不止。
少数患者以此为首发症状。
出血可持续数小时甚至数周。
出血程度与血浆因子活性(浓度)相关。
虽然正常止血所需的因子Ⅶ或Ⅸ的活性为25%,但有症状者其因子活性往往低于5%。
临床上依据因子活性将血友病分为重型、中型、轻型及亚临床型。
出血部位以四肢易受伤处最多见,可出现深部组织血肿,血肿大者可压迫附近的神经如:股神经、正中神经、尺神经引起疼痛及麻痹症状;压迫血管可发生坏疸。
颈部、喉部软组织出血可因呼吸道阻塞而窒息。
腹膜后、肠系膜出血可有腹痛。
重症者可出现鼻衄、牙龈出血、胃肠道出血、血尿,出血过多者可引起贫血。
关节腔反复出血见于重症患者,多发生在轻微损伤后,亦可自发出血。
可有局部肿胀、疼痛、压痛、急性症状持续3~5天,出血停止后约经数周积血逐渐吸收可不留痕迹。
凝血功能障碍凝血功能障碍是一种常见的疾病,其特征是患者在出血时凝血时间延长或凝血能力下降。
凝血功能障碍可由多种原因引起,包括先天性因素、遗传因素、疾病或药物引起的因素。
本文将讨论凝血功能障碍的病因、临床表现、诊断和治疗。
病因方面,凝血功能障碍可由于凝血因子缺乏或异常引起。
凝血因子是血浆中的蛋白质,参与血液凝固过程中的不同环节。
当某些凝血因子缺乏或受损时,凝血时间延长,导致出血时间延长。
常见的凝血因子缺乏病因包括遗传性血友病、先天性纤维蛋白原缺乏症等。
此外,一些疾病(如肝功能衰竭、肾功能衰竭)、药物(如抗凝剂、化疗药物)也可能影响凝血功能。
临床表现方面,凝血功能障碍的症状可因疾病严重程度和患者个体差异而有所不同。
常见的症状包括皮肤出血、黏膜出血、鼻衄、牙龈出血等。
病情严重的患者可能出现内脏出血、关节出血等较严重的并发症。
诊断方面,凝血功能障碍的诊断主要依靠实验室检查。
凝血功能检查主要包括凝血时间测定、凝血酶原时间测定、凝血酶活度测定等。
这些检查能够帮助医生确定凝血功能是否异常,并确定具体凝血因子的缺乏或异常。
治疗方面,针对凝血功能障碍的治疗主要包括凝血因子补充、抗凝剂的使用和其他辅助治疗等。
对于凝血因子缺乏的患者,可以通过注射凝血因子浓缩物来补充缺乏的凝血因子。
同时,对于存在凝血酶形成的风险的患者,可以使用抗凝剂来预防凝血。
此外,鼻腔或牙龈出血可以通过局部止血药物或外科手术来控制。
总之,凝血功能障碍是一种常见的疾病,可由多种原因引起。
基于细致的病史询问和实验室检查,可以进行准确的诊断。
针对不同病因,采取相应的治疗措施,可以有效地控制凝血功能障碍的症状,并避免严重的并发症的发生。
第十六章凝血障碍性疾病凝血障碍性疾病是凝血因子缺乏或功能异常所致的出血性疾病。
凝血障碍性疾病大致可分为先天性和获得性两类。
前者与生俱来,多为单一性凝血因子缺损,如血友病等;后者发病于出生后,常存在明显的基础疾病,多为复合性凝血因子减少,如肝病性出血等。
第一节血友病血友病(hemophilia)是一组因遗传性凝血活酶生成障碍引起的出血性疾病,包括血友病A、血友病B及遗传性FⅪ缺乏症,其中以血友病A最为常见。
血友病以阳性家族史、幼年发病、自发或轻度外伤后出血不止、血肿形成及关节出血为特征。
血友病的社会人群发病率为5~10/10万,婴儿发生率约1/5000。
血友病A、B及遗传性FⅪ缺乏的比较发病率为16:3:1,我国的血友病中,血友病A约占80%,血友病B约占15%,遗传性FⅪ缺乏症则极少见。
【病因与遗传规律】(一)病因血友病A又称遗传性抗血友病球蛋白缺乏症或FⅧ:C缺乏症。
FⅧ由两部分组成:即。
FWⅢ凝血活性部分(FⅧ:C)和vWI)因子(vWF)。
两者以复合物形式存在于血浆中。
前者被激活后参与FX的内源性激活;后者作为一种黏附分子参与血小板与受损血管内皮的黏附,并有稳定及保护FVⅢ:C的作用。
FⅧ:C基因位于X染色体长臂末端(Xq28),当其因遗传或突变而出现缺陷时,人体不能合成足量的FⅧ:C,导致内源性途径凝血障碍及出血倾向的发生。
血友病B又称遗传性FⅨ缺乏症。
FⅨ为一种单链糖蛋白,被Ⅺa等激活后参与内源性FX的激活。
FⅨ基因位于X染色体长臂末端(Xq26-q)。
遗传或突变使之缺陷时,不能合成足够量的FⅨ,造成内源性途径凝血障碍及出血倾向。
遗传性FⅪ缺乏症又称Rosenthal综合征。
(二)遗传规律血友病A、B均属X连锁隐性遗传性疾病。
其遗传规律见图6—16—1。
遗传性FⅪ缺乏症为常染色体隐性遗传性疾病,双亲都可遗传,子女均能发病。
【临床表现】(一)出血出血的轻重与血发病类型及相关因子缺乏程度有关。
血发病A出血较重,皿友病B则较轻。
按血浆FⅧ:C的活性,可将血友病A分为3型:①重型:F Ⅷ:C活性低于健康人的1%;②中型:FⅧ:c活性相当于健康人的1%~5%;③轻型:FⅧ:C活性相当于健康人的5%~25%。
血友病的出血多为自发性或轻度外伤、小手术后(如拔牙、扁桃体切除)出血不止,且具备下列特征:①生来俱有,伴随终身,但罕有出生时脐带出血;②常表现为软组织或深部肌肉内血肿;③负重关节如膝、踝关节等反复出血甚为突出,最终可致关节肿胀、僵硬、畸形,可伴骨质疏松、关节骨化及相应肌肉萎缩(血友病关节)。
重症患者可发生呕血、咯血,甚至颅内出血。
但皮肤紫癜罕见。
(二)血肿压迫症状及体征血肿压迫周围神经可致局部疼痛、麻木及肌肉萎缩;压迫血管可致相应供血部位缺血性坏死或淤血、水肿;口腔底部、咽后壁、喉及颈部出血可致呼吸困难甚至窒息;压迫输尿管致排尿障碍。
【实验室检查】(一)筛选试验CT正常或延长,APTT延长、凝血酶原消耗不良及简易凝血活酶生成试验(sTGT)异常,有助于血友病A的诊断及分型(表6—16—1)。
(二)确诊试验通过凝血活酶生成试验(TGT)及纠正试验,可确定3种血友病的诊断与鉴别诊断,见表6—16—2。
(三)特殊检查临床上,上述检测已可满足血友病的诊断要求,但对某些特殊病例或鉴定携带者,尚需进行下列特殊实验室检测:1.FⅧ:C、FⅪ抗原及活性测定;2.vWF抗原(vWFAg)测定;3.基因诊断。
【诊断与鉴别诊断】(一)诊断参考标准1.血友病A(1)临床表现:①男性患者,有或无家族史,有家族史者符合X连锁隐性遗传规律;②关节、肌肉、深部组织出血,可呈自发性,或发生于轻度损伤、小型手术后,易引起血肿及关节畸形。
(2)实验室检查:①CT正常或延长;②APTT多数延长,PCT、STGT多数异常;③。
TGT异常,并能被钡吸附正常血浆纠正;④FⅧ:C水平明显低下;⑤vWFAg 正常,FⅧ:c/vWFAg比值降低。
2.血友病B(1)临床表现:基本同血友病A,但程度较轻。
(2)实验室检查:①APTT延长,PCT缩短;②TGT延长,不能被钡吸附正常血浆纠正;③FⅨ抗原及活性明显减低。
3.遗传性因子Ⅺ缺乏症本病国内极少见,诊断标准从略。
4.携带者及胎儿产前诊断采用FⅧ:C、FⅨ定量检测、PCR及基因芯片技术等,可对携带者及胎儿作出诊断,以利优生优育。
(二)鉴别诊断主要应与血管性血友病鉴别,见本章第二节。
【治疗与预防】(一)一般治疗止血处理见本篇第十四章。
(二)替代疗法目前血友病的治疗仍以替代疗法为主,即补充缺失的凝血因子,它是防治血友病出血最重要的措施。
主要制剂有新鲜冷冻血浆(含所有的凝血因子)、冷沉淀物(主要含FⅧ、XⅢ、vWF及纤维蛋白原等,但FⅧ浓度较血浆高5~lO倍)、凝血酶原复合物(含FX、Ⅸ、Ⅶ、Ⅱ)、FⅧ浓缩制剂,或基因重组的纯化FⅧ等。
FⅧ:C及FⅨ的半衰期分别为8~12小时及18~30小时,故补充FⅧ需连续静脉滴注或每日2次;FⅨ每日1次即可。
FⅧ:C及FⅨ剂量:按每ml新鲜血浆含FⅧ或FⅨ1Iu计算,每输入1m1/kg血浆,可提高患者FⅧ:C或FⅨ水平2%。
最低止血要求FⅧ:C或FⅨ水平达20%以上,出血严重或欲行中型以上手术者,应使FⅧ或FⅨ活性水平达40%以上。
凝血因子的补充一般可采取下列公式计算:首次输入FⅧ:C(或FⅨ)剂量(IU)=体重×所需提高的活性水平(%)÷2 重组人活化因子Ⅶ(rFⅦa)可用于防治产生了FⅧ或FⅨ抗体的血友病患者的出血,但有增加血栓形成的副作用。
常用剂量是90μg/kg,每2~3小时静脉注射,直至出血停止。
(三)药物治疗1.去氨加压素(desmopressin,DDAVP) 此药作用见本篇第十四章。
常用剂量为16~32μg/次,置于30ml生理盐水内快速滴入,每12小时1次。
亦可分次皮下注射或鼻腔滴入。
2.达那唑(danazo1) 300~600mg/d,顿服或分次口服,对轻、中型者疗效较好,其作用机制不明。
3.糖皮质激素通过改善血管通透性及减少抗FⅧ:C抗体的产生而发挥作用。
适用于反复接受FⅧ:C输注治疗而疗效渐差的患者。
4.抗纤溶药物通过保护已形成的纤维蛋白凝块不被溶解而发挥止血作用。
药物种类见本篇第十四章。
5.家庭治疗血友病患者的家庭治疗在国外已广泛应用。
除有抗FⅧ:C 抗体、病情不稳定、小于3岁的患儿外,均可安排家庭治疗。
血友病患者及其家属应接受有关疾病的病理、生理、诊断及治疗知识的教育,家庭治疗最初应在专业医师的指导下进行。
除传授注射技术外,还包括血液病学、矫形外科、精神、心理学以及艾滋病、病毒性肝炎的预防知识等。
(四)外科治疗有关节出血者应在替代治疗的同时,进行固定及理疗等处理。
对反复关节出血而致关节强直及畸形的患者,可在补充足量FⅧ:C或FⅨ的前提下,行关节成型或人工关节置换术。
(五)基因疗法现正在研究将决定FⅧ:C、FⅨ及FⅪ合成的正常基因,通过载体以直接或间接方式转导人患者体内的方法,以纠正血友病的基因缺陷,生成足够的FⅧ:C、FⅨ或FⅪ。
(六)预防由于本病目前尚无根治方法,因此预防更为重要。
血友病的出血多数与损伤有关,预防损伤是防止出血的重要措施之一,医务人员应向患者家属、学校、工作单位及本人介绍有关血友病出血的预防知识。
对活动性出血的患者,应限制其活动范围和活动强度。
一般血友病患者,应避免剧烈或易致损伤的活动、运动及工作,减少出血的危险;建立遗传咨询,严格婚前检查,加强产前诊断,是减少血友病发生的重要方法。
第二节血管性血友病血管性血友病(yon willebrand disease,vWD)分为两种:遗传性血管性血友病(cVWD)和获得性血管性血友病(avWD)。
遗传性血管性血友病是一种常染色体遗传性疾病,多为显性遗传。
以自幼发生的出血倾向、出血时间延长、血小板黏附性降低、瑞斯托霉素诱导的血小板聚集缺陷,及血浆vWFAg缺乏或结构异常为特点。
在遗传性出血性疾病中,其发病率可能仅次于血友病,约为4~10/10万,但在我国本病的发生率较低。
获得性血管性血友病可在多种疾病的基础上发生,少数患者可无基础疾病。
【病因与发病机制】vWF主要存在于内皮细胞、巨核细胞及血小板,生理功能:①与FⅧ:C以非共价键结合成vWF-FⅧ:C复合物,即FⅧ。
vWF增加FⅧ:C稳定性、防止其降解,并促进其生成及释放;②vWF在血小板与血管壁的结合中起着重要的桥梁作用。
血小板活化时,vWF的一端与血小板糖蛋白I b结合,另一端则与受损伤血管壁的纤维结合蛋白及胶原结合,使血小板能牢固地黏附于血管内皮;③vWF 可与血小板糖蛋白Ⅱb/Ⅲa结合,诱导血小板聚集。
vWF基因位于12号染色体短臂末端,当其缺陷时,vWF生成减少或功能异常,伴随FⅧ:C中度减低,血小板黏附、聚集功能障碍。
获得性血管性血友病涉及多种发病机制。
最常见的是产生具有抗vWF活性的抑制物,主要为IgG;其次为肿瘤细胞吸附vWF,使血浆vWF减少;另外,抑制物可与vWF的非活性部位结合形成复合物,加速其在单核一巨噬细胞系统的破坏。
【临床表现】出血倾向是本病的突出表现。
与血友病比较,其出血在临床上有以下特征:①出血以皮肤黏膜为主,如鼻出血、牙龈出血、瘀斑等,外伤或小手术(如拔牙)后的出血也较常见;②男女均可发病,女性青春期患者可有月经过多及分娩后大出血;③出血可随年龄增长而减轻,此可能与随着年龄增长而vWF活性增高有关;④自发性关节、肌肉出血相对少见,由此致残者亦少。
【实验室检查】(一)出血时间BT延长是vWD最常见的实验室异常,阿司匹林耐量试验多呈阳性。
口服阿司匹林O.6g后2小时及4小时测BT,比服药前延长2分钟以上为阳性。
(二)血小板黏附试验多数患者血小板黏附功能减低。
(三)瑞斯托霉素血小板聚集试验(RlPA)患者血小板对瑞斯托霉素的诱导不产生聚集,为vWD的特异性诊断试验之一。
(四)vWF抗原(vWFAg)测定在多数vWD患者降低。
(五)FⅧ:C活性测定多数患者FⅧ:C活性中度降低。
【诊断与分型】(一)诊断要点①有或无家族史,有家族史者多数符合常染色体显性遗传规律;②血小板计数和形态正常,出血时间延长或阿司匹林耐量试验阳性,血小板黏附功能减低或正常,瑞斯托霉素诱导血小板聚集障碍;③vWFAg、FⅧ:C活性减低;④排除血小板功能缺陷性疾病。
(二)鉴别诊断本病根据阿司匹林耐量试验和vWFAg测定可与血友病A、B鉴别,根据血小板形态可与巨血小板综合征鉴别。
(三)分型根据遗传方式、临床表现、实验室检查特别是分子生物学分析,可将vWD 分为下列常见类型(表6—16—3)。
【治疗】(一)一般治疗同血友病。
【二)替代治疗1.制剂新鲜冷冻血浆及冷沉淀物、FⅧ浓缩制剂等,均含有vWF,适量补充可有效提高vWF、水平,多数制剂还可同时补充FⅧ:C,有良好的治疗作用。