可疑医疗器械不良事件报告表和例子word正常版
- 格式:pdf
- 大小:41.88 KB
- 文档页数:3
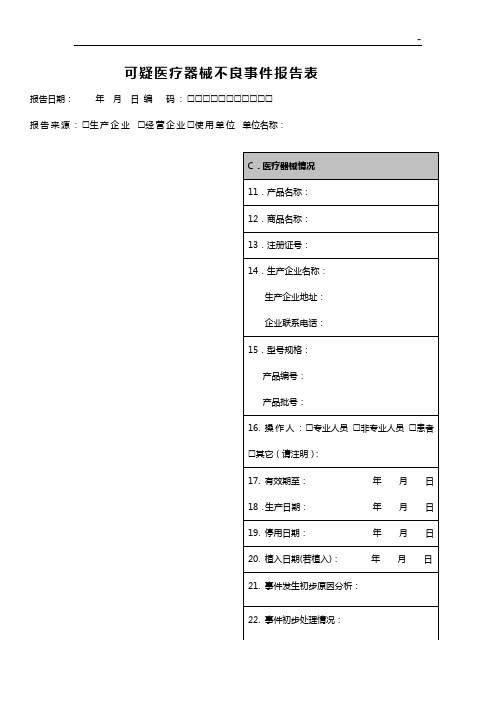

济宁市第一人民医院西院区可疑医疗器械不良事件报告表报告日期:年月日编码:报告来源:生产企业经营企业使用单位单位名称:联系地址:邮编:联系电话:A.患者资料C.医疗器械情况1.姓名:2.年龄:3.性别男女4.预期治疗疾病或作用:B.不良事件情况5.事件主要表现:6.事件发生日期:年月日7.发现或者知悉时间:年月日8.医疗器械实际使用场所:医疗机构家庭其它(请注明):9.事件后果死亡(时间);危及生命;机体功能结构永久性损伤;可能导致机体功能机构永久性损伤;需要内、外科治疗避免上述永久损伤;其它(在事件陈述中说明)。
10.事件陈述:(至少包括器械使用时间、使用目的、使用依据、使用情况、出现的不良事件情况、对受害者影响、采取的治疗措施、器械联合使用情况)报告人:医师技师护士其他报告人签名:11.产品名称:12.商品名称:13.注册证号:14.生产企业名称:生产企业地址:企业联系电话:15.型号规格:产品编号:产品批号:16.操作人:专业人员非专业人员患者其它(请注明):17.有效期至:年月日18.生产日期:年月日19.停用日期:年月日20.植入日期(若植入):年月日21.事件发生初步原因分析:22.事件初步处理情况:23.事件报告状态:已通知使用单位已通知生产企业已通知经营企业已通知药监部门D.不良事件评价24.省级监测技术机构评价意见(可另附附页):25.国家监测技术机构评价意见(可另附附页):国家食品药品监督管理局制医疗器械不良事件监测基础知识问答1.审批上市的医疗器械都是绝对安全的吗?不是。
任何医疗器械产品都具有一定的使用风险,都可能因为当时科技水平的制约、实验条件的限制等因素,而在临床应用过程中存在一定的风险。
所谓批准上市,是指社会、技术、伦理和法令皆可接受的基础上的认可,而非绝对安全。
被批准上市的医疗器械只是“效益大于风险”的“风险可接受”产品,即被批准上市产品在现有认识水平下,相对符合安全使用的要求。

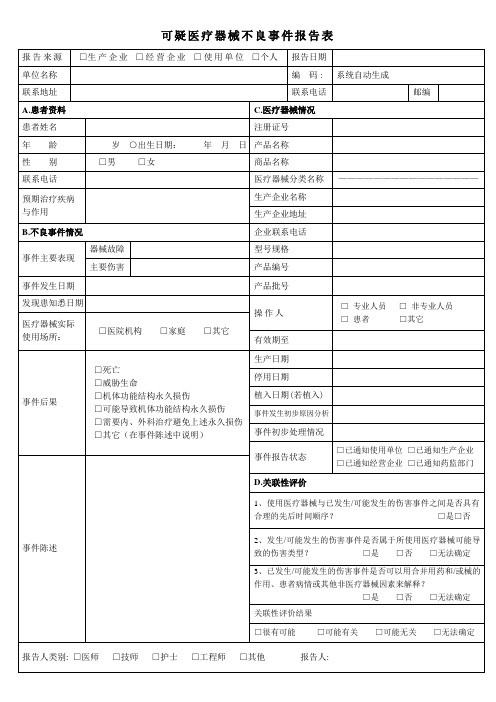



7可疑医疗器械不良事件报告表报告日期:____年__月__日编码:报告来源:生产企业经营企业使用单位单位名称:联系地址:邮编:联系电话:报告人签名: 填写要求:《可疑医疗器械不良事件报告表》由题眉、患者资料、不良事件情况、医疗器械情况、关联性评价、不良事件评价及题末7部分组成。
1.题眉A .报告日期:是指填报人填报该次不良事件时的确切时间。
B .编码:由省(区、市)医疗器械不良事件监测技术机构填写,按以下排列方式:省 (区、市) 年份流水号 □□□□□□□□□□□报告人: 医师 技师 护士 其他注:省(区、市)编码按中华人民共和国行政区划代码填写。
在医疗器械不良事件监测系统中,编码由系统自动生成。
C.报告来源:是指填报可疑医疗器械不良事件单位的类别,填写时请选择相应的选项,并在“□”中划“√”。
D.单位名称:是指填报可疑医疗器械不良事件单位的全称,不可用简称。
E.联系地址、电话及邮编:是指填报可疑医疗器械不良事件单位的联系地址、电话及邮编。
2.患者资料A.患者姓名:是指患者真实全名。
若患者姓名无法获知,应填写未知;新生儿无姓名,应填写××子或××女。
B.年龄:是指患者发生可疑医疗器械不良事件时的实际年龄,字体为阿拉伯数字。
若患者年龄小于1岁,应填写具体的月份或天数,如6个月。
C.性别:是指患者的性别,填写时请选择相应的选项,并在“□”中划“√”。
D.预期治疗疾病或作用:是指涉及不良事件的医疗器械用于治疗的疾病或者预计使用该医疗器械所发挥的作用,例如血管内支架用于治疗急性心肌梗死。
3.不良事件情况A.事件主要表现:是指使用医疗器械后引发的、可能与该医疗器械使用有关的有害事件(且与质量、医疗事故无关)。
填写不良事件主要表现要明确、具体,如放臵节育器后,出现意外脱落者,可填写“节育器脱落”。
B.事件发生日期:是指不良事件发生的确切时间,如:×年×月×日,字体为阿拉伯数字。

附件1:可疑医疗器械不良事件报告表报告日期: 年 月 日 编 码: 报告来源: 生产企业 经营企业 使用单位 单位名称:联系地址: 邮 编: 联系电话:报告人签名:报告人: 医师 技师 护士 其他填写要求《可疑医疗器械不良事件报告表》由题眉、患者资料、不良事件情况、医疗器械情况、关联性评价、不良事件评价及题末7部分组成。
1.题眉A.报告日期:是指填报人填报该次不良事件时的确切时间。
B.编码:由省(区、市)医疗器械不良事件监测技术机构填写,按以下排列方式:省(区、市)年份流水号□□□□□□□□□□□注:省(区、市)编码按中华人民共和国行政区划代码填写。
在医疗器械不良事件监测系统中,编码由系统自动生成。
C.报告来源:是指填报可疑医疗器械不良事件单位的类别,填写时请选择相应的选项,并在“□”中划“√”。
D.单位名称:是指填报可疑医疗器械不良事件单位的全称,不可用简称。
E.联系地址、电话及邮编:是指填报可疑医疗器械不良事件单位的联系地址、电话及邮编。
2.患者资料A.患者姓名:是指患者真实全名。
若患者姓名无法获知,应填写未知;新生儿无姓名,应填写××子或××女。
B.年龄:是指患者发生可疑医疗器械不良事件时的实际年龄,字体为阿拉伯数字。
若患者年龄小于1岁,应填写具体的月份或天数,如6个月。
C.性别:是指患者的性别,填写时请选择相应的选项,并在“□”中划“√”。
D.预期治疗疾病或作用:是指涉及不良事件的医疗器械用于治疗的疾病或者预计使用该医疗器械所发挥的作用,例如血管内支架用于治疗急性心肌梗死。
3.不良事件情况A.事件主要表现:是指使用医疗器械后引发的、可能与该医疗器械使用有关的有害事件(且与质量、医疗事故无关)。
填写不良事件主要表现要明确、具体,如放臵节育器后,出现意外脱落者,可填写“节育器脱落”。
B.事件发生日期:是指不良事件发生的确切时间,如:×年×月×日,字体为阿拉伯数字。

附件1:国家食品药品监督管理局制可疑医疗器械不良事件报告表报告日期:年月日编码:报告来源:生产企业经营企业使用单位单位名称:联系地址: 邮编:联系电话:A.患者资料1.姓名:2.年龄: 3.性别男女4.预期治疗疾病或作用:B.不良事件情况5.事件主要表现:6.事件发生日期:年月日7.发现或者知悉时间:年月日8. 医疗器械实际使用场所:医疗机构家庭其它(请注明):9.事件后果死亡(时间);危及生命;机体功能结构永久性损伤;可能导致机体功能机构永久性损伤;需要内、外科治疗避免上述永久损伤;其它(在事件陈述中说明)。
例子:附件1:国家食品药品监督管理局制可疑医疗器械不良事件报告表报告日期:2010年10月25日 编 码:报告来源:生产企业经营企业√使用单位 单位名称:按实际填写10.事件陈述:(至少包括器械使用时间、使用目的、使用依据、使用情况、出现的不良事件情况、对受害者影响、采取的治疗措施、器械联合使用情况)报告人: 医师 技师 护士 其他 报告人签名:C .医疗器械情况11.产品名称: 12.商品名称: 13.注册证号: 14.生产企业名称:生产企业地址: 企业联系电话: 15.型号规格:产品编号: 产品批号:16. 操作人:专业人员 非专业人员 患者其它(请注明):17. 有效期至: 年 月 日18.生产日期: 年 月 日 19. 停用日期: 年 月 日20. 植入日期(若植入): 年 月 日 21. 事件发生初步原因分析:22. 事件初步处理情况:23.事件报告状态:已通知使用单位 已通知生产企业 已通知经营企业已通知药监部门D. 不良事件评价24.省级监测技术机构评价意见(可另附附页):25.国家监测技术机构评价意见(可另附附页): C .医疗器械情况11.产品名称:一次性使用静脉留置针12.商品名称:13.注册证号:国食药监械(准)字2007第3150968号联系地址: 按实际填写 邮编: 联系电话:16. 操作人:√专业人员非专业人员患者 其它(请注明):23.事件报告状态: 已通知使用单位 已通知生产企业 已通知经营企业已通知药监部门3.性别男女√8. 医疗器械实际使用场所:√ 医疗机构 家庭 其它(请注明): 9.事件后果死亡 (时间); 危及生命;机体功能结构永久性损伤; 可能导致机体功能机构永久性损伤; 需要内、外科治疗避免上述永久损伤; √ 其它(在事件陈述中说明)。


7可疑医疗器械不良事件报告表报告日期:____年__月__日编码:报告来源:生产企业经营企业使用单位单位名称:联系地址:邮编:联系电话:报告人签名: 填写要求:《可疑医疗器械不良事件报告表》由题眉、患者资料、不良事件情况、医疗器械情况、关联性评价、不良事件评价及题末7部分组成。
1.题眉A .报告日期:是指填报人填报该次不良事件时的确切时间。
B .编码:由省(区、市)医疗器械不良事件监测技术机构填写,按以下排列方式:省 (区、市) 年份流水号 □□□□□□□□□□□报告人: 医师 技师 护士 其他注:省(区、市)编码按中华人民共和国行政区划代码填写。
在医疗器械不良事件监测系统中,编码由系统自动生成。
C.报告来源:是指填报可疑医疗器械不良事件单位的类别,填写时请选择相应的选项,并在“□”中划“√”。
D.单位名称:是指填报可疑医疗器械不良事件单位的全称,不可用简称。
E.联系地址、电话及邮编:是指填报可疑医疗器械不良事件单位的联系地址、电话及邮编。
2.患者资料A.患者姓名:是指患者真实全名。
若患者姓名无法获知,应填写未知;新生儿无姓名,应填写××子或××女。
B.年龄:是指患者发生可疑医疗器械不良事件时的实际年龄,字体为阿拉伯数字。
若患者年龄小于1岁,应填写具体的月份或天数,如6个月。
C.性别:是指患者的性别,填写时请选择相应的选项,并在“□”中划“√”。
D.预期治疗疾病或作用:是指涉及不良事件的医疗器械用于治疗的疾病或者预计使用该医疗器械所发挥的作用,例如血管内支架用于治疗急性心肌梗死。
3.不良事件情况A.事件主要表现:是指使用医疗器械后引发的、可能与该医疗器械使用有关的有害事件(且与质量、医疗事故无关)。
填写不良事件主要表现要明确、具体,如放臵节育器后,出现意外脱落者,可填写“节育器脱落”。
B.事件发生日期:是指不良事件发生的确切时间,如:×年×月×日,字体为阿拉伯数字。



可疑医疗器械不良事件报告表
报告日期: 年 月 日 编 码: 报告来源: 生产企业 经营企业 使用单位 单位名称:
联系地址: 邮 编: 联系电话:
报告人签名: 国家食品药品监督管理局制
医疗器械不良事件补充报告表
报告人: 医师 技师 护士 其他
报告时间:年月日编码:
报告人: 省级监测技术机构接收日期: 国家监测技术机构接收日期:
生产企业(签章)
国家食品药品监督管理局制
医疗器械不良事件年度汇总报告表
报告时间:年月日编码:
汇总时间:年月日至年月日
报告人: 省级监测技术机构接收日期:国家监测技术机构接收日期:
生产企业(签章)
国家食品药品监督管理局制
附件4:
市级监测机构年度考核评分表
注:评分表总分值为100分,满70分为合格。
生产、经营企业MDR监测工作检查评分表
注:评分表总分值为100分,满70分为合格。

附件1:(一)
国家食品药品监督管理局制
可疑医疗器械不良事件报告表
报告日期:年月日编码:
报告来源: 生产企业 经营企业 使用单位单位名称:
联系地址: 邮编:联系电话:
报告人签名:
例子:
附件1: 国家食品药品监督管理局制
可疑医疗器械不良事件报告表
报告日期:10月25日 编 码:
报告来源: 生产企业 经营企业 √使用单位 单位名称:按实际填写
联系地址: 按实际填写 邮 编: 联系电话:
附件
1:
国家食品药品监督管理局制
可疑医疗器械不良事件报告表
报告日期: 2010 年 10 月 8日 编 码:
报告人签名:
报告来源: 生产企业 经营企业 √使用单位单位名称:联系地址: 邮编:联系电话:
报告人签名:。

可疑医疗器械不良事件报告表
报告日期: 年 月 日 编 码:? ? ? ? ? ? ? ? ? ? ? 报告来源:? 生产企业 ? 经营企业 ? 使用单位 单位名称:
联系地址: 邮 编: 联系电话:
报告人签名: 国家食品药品监督管理局制
医疗器械不良事件补充报告表
报告人: 医师? 技师? 护士? 其他?
报告时间:年月日编码:? ? ? ? ? ? ? ? ? ? ?
报告人: 省级监测技术机构接收日期: 国家监测技术机构接收日期:
生产企业(签章)
国家食品药品监督管理局制
医疗器械不良事件年度汇总报告表
报告时间:年月日编码:? ? ? ? ? ? ? ? ? ? ?
汇总时间:年月日至年月日
报告人: 省级监测技术机构接收日期:国家监测技术机构接收日期:
生产企业(签章)
国家食品药品监督管理局制
附件4:
市级监测机构年度考核评分表
注:评分表总分值为100分,满70分为合格。
生产、经营企业MDR监测工作检查评分表
注:评分表总分值为100分,满70分为合格。
注:评分表总分值为100分,满70分为合格。


医疗器械不良事件报告范例近年来,随着社会经济的发展和医疗技术的突破,医疗器械在医疗行业中的应用越来越广泛。
然而,与之同时,医疗器械不良事件也时有发生,给人们的生命安全带来了一定的威胁。
为了更好地管理和控制医疗器械不良事件,必须建立一个完善的报告系统。
本文将以一个医疗器械不良事件的报告范例为例,详细介绍该报告的内容和形式。
报告主题:医用电动吸引器失灵一、事件描述根据我院2021年5月1日至5月30日期间发生的医疗器械不良事件报告,报告编号:12345678。
事发时间为2021年5月15日上午10点,患者王某(男,65岁)在我院进行鼻窦炎手术后出现呼吸困难的不良状况。
经过紧急救治,患者目前病情稳定。
二、器械信息患者在手术时使用了我院购买的医用电动吸引器丙型型号(批次号:ABC123456)。
该器械主要用于排除呼吸道分泌物,确保患者的通畅呼吸。
三、失灵情况描述据手术室护士陈某的描述,手术期间,电动吸引器突然停止运转,无法完成抽吸任务。
护士随即通知同事切换到备用吸引器继续手术。
经过后续的检查和调查,确定该电动吸引器发生了严重故障,导致了此次意外。
四、结果由于电动吸引器故障,患者出现呼吸困难。
幸好我们的医护人员反应及时,迅速采取了紧急救治措施,患者目前病情稳定。
五、原因分析根据技术人员的分析,初步判断电动吸引器失灵的原因是设备内部的电线连接问题,导致电路中断,使得设备无法正常运行。
六、整改措施1. 停止使用该型号的电动吸引器,并将其送至供应商修理。
2. 加强器械设备的定期检查和维护,确保设备的正常运行。
3. 设立医疗器械不良事件报告制度,加强对医疗器械的质量监管。
七、教训与总结本次医疗器械不良事件表明,医疗机构在选择和使用医疗器械时必须十分谨慎。
对于设备故障要及时发现和排除,并加强对设备的定期检查和维护。
同时,建立医疗器械不良事件报告制度,及时报告和处理医疗器械不良事件,以提高患者的安全性和医疗服务质量。
附件1:国家食品药品监督管理局制
可疑医疗器械不良事件报告表
报告日期:年月日编码:
报告来源:生产企业经营企业使用单位单位名称:
联系地址: 邮编:联系电话:
A.患者资料
1.姓名:2.年龄: 3.性别男女4.预期治疗疾病或作用:
B.不良事件情况
5.事件主要表现:
6.事件发生日期:年月日
7.发现或者知悉时间:年月日
8. 医疗器械实际使用场所:
医疗机构家庭其它(请注明):
9.事件后果
死亡(时间);
危及生命;
机体功能结构永久性损伤;
可能导致机体功能机构永久性损伤;
需要内、外科治疗避免上述永久损伤;
其它(在事件陈述中说明)。
10.事件陈述:(至少包括器械使用时间、使用目的、使
用依据、使用情况、出现的不良事件情况、对受害者影
响、采取的治疗措施、器械联合使用情况)
报告人:医师技师护士其他
报告人签名:C.医疗器械情况
11.产品名称:
12.商品名称:
13.注册证号:
14.生产企业名称:
生产企业地址:
企业联系电话:
15.型号规格:
产品编号:
产品批号:
16. 操作人:专业人员非专业人员患者其它(请注明):
17. 有效期至:年月日18.生产日期:年月日
19. 停用日期:年月日
20. 植入日期(若植入):年月日
21. 事件发生初步原因分析:
22. 事件初步处理情况:
23.事件报告状态:
已通知使用单位已通知生产企业
已通知经营企业已通知药监部门
D. 不良事件评价
24.省级监测技术机构评价意见(可另附附页): 25.国家监测技术机构评价意见(可另附附页):
例子:
附件1:
国家食品药品监督管理局制
可疑医疗器械不良事件报告表
报告日期:2010年10月25日编码:
报告来源:
生产企业
经营企业
√使用单位
单位名称:按实际填写
联系地址: 按实际填写
邮
编:
联系电话:
C .医疗器械情况
11.产品名称:一次性使用静脉留置针12.商品名称:
13.注册证号:国食药监械(准)字2007第3150968号
14.生产企业名称:
江西洪达医疗器械集团有限公司
生产企业地址:南昌市进贤县城胜利南路39号
企业联系电话:0791-562888815.型号规格:24GA
产品编号:201005003601 产品批号:10050216. 操作人:√专业人员
非专业人员
患者
其它(请注明):
17. 有效期至:2013 年 04 月 30 日18.生产日期:
2010年 05月 02 日
19. 停用日期: 2010年 10月 22 日20. 植入日期(若植入): 2010 年 10 月20日
21. 事件发生初步原因分析:部分患者对一次性使
用静脉留置针发生排斥反应而致穿刺部位组织红
肿。
22. 事件初步处理情况:拔除一次性使用静脉留置
针
23.事件报告状态:
已通知使用单位已通知生产企业已通知经营企业
已通知药监部门
D. 不良事件评价
24.省级监测技术机构评价意见(可另附附页)
:
25.国家监测技术机构评价意见(可另附附页):
A .患者资料
1.姓名:
2.年龄: 3.性别
男
女√
4.预期治疗疾病或作用
:输液
B .不良事件情况
5.事件主要表现:穿刺部位红肿6.事件发生日期:2010 年 10 月 22 日7.发现或者知悉时间: 2010 年 10 月 22 日
8. 医疗器械实际使用场所:√医疗机构家庭
其它(请注明):9.事件后果
死亡(时间);
危及生命;
机体功能结构永久性损伤;可能导致机体功能机构永久性损伤;需要内、外科治疗避免上述永久损伤;√其它(在事件陈述中说明)。
10.事件陈述:(至少包括器械使用时间、使用目的、使用依据、使用情况、出现的不良事件情况、对受害者影响、采取的治疗措施、器械联合使用情况)
患者于2010年10月20日在我院留医治疗使用一次性使用静脉留置针,穿刺部位于穿刺输液后2天出现皮
肤红肿,无发热等现象,拔除一次性使用静脉留置针
1
天后症状消失。
报告人:医师√技师护士其他
报告人签名:
例子 2
附件1:国家食品药品监督管理局制
可疑医疗器械不良事件报告表
报告日期: 2010 年 10 月 8日编码:
报告来源:生产企业经营企业√使用单位单位名称:
联系地址:邮编:联系电话:A.患者资料
1.姓名:黄国兆2.年龄:62 3.性别√男女
4.预期治疗疾病或作用:监护病人生命体征变化B.不良事件情况
5.事件主要表现:监护仪黑屏
6.事件发生日期:2010 年 10月 6日
7.发现或者知悉时间:2010 年 10月 6日
8. 医疗器械实际使用场所:
√医疗机构家庭其它(请注明):
9.事件后果
死亡(时间);
危及生命;
机体功能结构永久性损伤;
可能导致机体功能机构永久性损伤;
需要内、外科治疗避免上述永久损伤;
√其它(在事件陈述中说明)。
10.事件陈述:(至少包括器械使用时间、使用目的、
使用依据、使用情况、出现的不良事件情况、对受
害者影响、采取的治疗措施、器械联合使用情况)患者因心脏病发作到我科治疗,于2010年10月6日10时20分使用多参数监护仪监护病人,至30分钟, 多参数监护仪显示屏突然黑屏,因而无法监测到病人生命体征,立即更换多参数监护仪,对患者没有造成不良影响。
报告人:医师技师护士√其他
报告人签名:C.医疗器械情况
11.产品名称:多参数监护仪
12.商品名称:
13.注册证号:粤食药监械(准)字2004第3210440号14.生产企业名称:深圳万瑞医疗电子股份有限公司生产企业地址:
企业联系电话:8008303312
15.型号规格:PM-9000
产品编号:
产品批号:03000171
16. 操作人:√专业人员非专业人员患者其它(请注明):
17. 有效期至:年月日18.生产日期:年月日
19. 停用日期: 2010年 10月 6日
20. 植入日期(若植入):年月日
21. 事件发生初步原因分析:多参数监护仪各部件接触不良.
22. 事件初步处理情况:更换多参数监护仪
23.事件报告状态:
已通知使用单位已通知生产企业
已通知经营企业已通知药监部门
D. 不良事件评价
24.省级监测技术机构评价意见(可另附附页): 25.国家监测技术机构评价意见(可另附附页):。