高乳酸血症相关遗传病诊断 -2
- 格式:ppt
- 大小:4.60 MB
- 文档页数:45
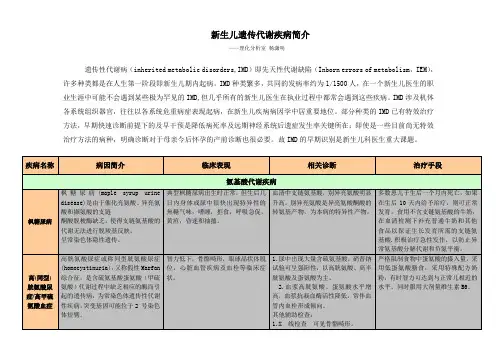
新生儿遗传代谢疾病简介——理化分析室杨潇明遗传性代谢病(inherited metabolic disorders,IMD)即先天性代谢缺陷(Inborn errors of metabolism,IEM),许多种类都是在人生第一阶段即新生儿期内起病。
IMD种类繁多,共同的发病率约为1/1500人,在一个新生儿医生的职业生涯中可能不会遇到某些极为罕见的IMD,但几乎所有的新生儿医生在执业过程中都常会遇到这些疾病。
IMD涉及机体各系统组织器官,往往以各系统危重病症表现起病,在新生儿疾病病因学中居重要地位。
部分种类的IMD已有特效治疗方法,早期快速诊断前提下的及早干预是降低病死率及远期神经系统后遗症发生率关键所在;即使是一些目前尚无特效治疗方法的病种,明确诊断对于母亲今后怀孕的产前诊断也很必要。
故IMD的早期识别是新生儿科医生重大课题。
性测定可进行产前诊断。
其他辅助检查:B 超可发现肝脏肿大,脑电图异常脑波,智力检查有智力水平落后等表现。
异戊二酸血症由于异戊二酸辅酶A去氢酶缺乏所致。
发病年龄为0~1岁,急性期症状包括呕吐,缺乏食欲,无精打采,嗜睡,神经症状,体温低。
通常发作原因为上呼吸道感染或摄取太多高蛋白食物。
异戊酸血症与U型戊二酸血症均可出现特征性的“汗脚臭”味是诊断这两种疾病依据之一。
异戊酸血症与U型戊二酸血症均可出现特征性的“汗脚臭”味是诊断这两种疾病依据之一。
尿液有机酸层析可持续地检测到异戊酞甘二酸.感染或大量进食蛋白质后,还可检测到3-经异戊酸。
本病给予低蛋白或低亮氨酸饮食及三种支链氨基酸饮食效果较好。
口服甘氨酸可以预防并发症,但需注意高甘氨酸血症所致的神经毒性反应.也可口服L-肉毒碱。
戊二酸血症Ⅰ型是一种胺基酸代谢异常的遗传疾病,主要是因为戊二基辅酶A去氢酶这个分解赖胺酸(lysine)与色胺酸以及羟赖胺酸代谢途径中的酵素有缺陷所导致,造成有毒的代谢中间产物戊二酸在人体内过量堆积,堆积血液与组织中且排泄于尿液,造成急性的代谢异常与渐进性的神经症状,影响患者的脑部组织,导致日后的生理发育障碍与智能迟缓等后遗症。


1例罕见线粒体病分析毕玫荣;王莹;徐谧;朱薇薇【摘要】线粒体疾病通常累及多个系统,表现具有高度差异,基因检测为线粒体疾病诊断的金标准。
本文中患儿为生后即以反应差、酸中毒、贫血发病,给予基因检测确诊为联合氧化磷酸化缺陷14型。
此病非常罕见,资料显示仅有两例报道。
其父母分别含有一个突变基因,且这两个突变均未见报道。
%Mitochondrial diseases usual y involve multiple organ systems,present with a greatness diversity of clinical manifestations,genetic testing is the gold standard for the diagnosis of mitochondrial diseases,here we present a case of disease,a baby with poor response,acidosis and anemia after born was final y diagnosed with the combined oxidative phosphorylation defect type 14 by genetic testing,the disease is rare and only two cases are reported, their parents each contain a mutated gene which is not present in current documents.【期刊名称】《中国卫生标准管理》【年(卷),期】2015(000)025【总页数】2页(P91-92)【关键词】线粒体病;酸中毒;基因检测【作者】毕玫荣;王莹;徐谧;朱薇薇【作者单位】250013山东大学附属济南市中心医院;250013山东大学附属济南市中心医院;250013山东大学附属济南市中心医院;250013山东大学附属济南市中心医院【正文语种】中文【中图分类】R5961例罕见线粒体病分析毕玫荣王莹徐谧朱薇薇【摘要】线粒体疾病通常累及多个系统,表现具有高度差异,基因检测为线粒体疾病诊断的金标准。




2021高乳酸血症的诊治思路(全文)乳酸是碳水化合物和非必需氨基酸代谢过程中重要的中间代谢产物之一,属强酸性物质,在体内几乎完全以离子化的乳酸根和氢离子状态存在。
临床测定的是血乳酸根浓度,并以此代表血乳酸浓度,正常值为0.5-1.5 mmol/L , > 2 mmol/L 为高乳酸血症,—般以2~5 mmol/L 为轻度高乳酸血症,> 5 mmol/L为重度高乳酸血症。
孚L酸酸中毒则指乳酸増高同时伴代谢性酸中毒,血pH < 7.35,伴阴离子间隙(anion gap , AG)增高,多见于重度高乳酸血症患者。
血乳酸水平增高常见于危重症患者,其增高幅度和住院病死率呈正相关,动态监测乳酸变化更有助评估预后。
儿童高乳酸血症原因复杂,结合临床特点尽快明确病因对及时诊治、改善预后至关重要。
-乳酸代谢葡萄糖无氧代谢是体内乳酸的最主要来源。
无氧糖酵解是葡萄糖代谢的第一步,在细胞质中完成,在多种酶作用下,葡萄糖先被分解为丙酮酸, 丙酮酸在乳酸脱氢酶(lactate dehydrogenase , LDH )作用下生成乳酸,乳酸也可逆向生成丙酮酸。
在有氧代谢情况下,丙酮酸进入线粒体内,经三竣酸循环实现葡萄糖的完全氧化,生成二氧化碳和水。
二氧化碳部分转化为碳酸氢盐维持体内酸碱平衡,部分经呼吸排出体外。
生糖氨基酸等糖异生底物通过糖异生作用生成葡萄糖,丙氨酸可在丙氨酸转氨酶作用下生成丙酮酸,参与乳酸的生成。
几乎身体的所有组织均可产生乳酸,肝脏和肾脏是摄取和利用乳酸的主要器官,当乳酸产生増多、摄取减少或两者同时存在时,则血乳酸水平增高。
LDH 有左旋LDH ( levorotatory LDH , L - LDH )和右旋LDH (dextrorotatory LDH , D - LDH )两种光学同分异构体。
人体内只有L-LDH ,只能生成和利用L -乳酸;某些细菌,如肠道内的乳酸杆菌、双歧杆菌等具有L - LDH和D - LDH ,可分别生成L -乳酸和D -乳酸,并通过肠黏膜吸收入血。

乳酸偏高游离肉碱偏低谱系随着人们生活水平的不断提高,对健康的关注度也越来越高。
健康是我们生活中不可或缺的一部分,而一些生化指标就是我们身体健康状况的重要体现。
本文将围绕乳酸偏高、游离肉碱偏低、谱系等主题展开讨论,以帮助读者更好地了解这些生化指标在我们身体健康中的作用。
一、乳酸偏高1.乳酸是一种重要的有机酸,它在我们的身体内扮演着重要的角色。
在正常情况下,乳酸的生成和清除是处于平衡状态的。
然而,一些疾病或生活方式不当可能导致乳酸偏高,从而成为健康的隐患。
2.乳酸偏高的原因多种多样,可能是由于缺氧、糖代谢障碍、肝功能不全等引起的。
当我们发现乳酸偏高时,要及时寻求专业医生的帮助,进行准确的诊断和治疗。
3.另外,乳酸偏高也可能是因为我们的生活方式不当所致。
长时间的剧烈运动、缺乏睡眠、饮食不均衡等都可能导致乳酸偏高,因此我们在日常生活中也要注意调整好自己的生活方式,预防乳酸偏高的发生。
二、游离肉碱偏低1.游离肉碱是一种重要的脂肪酸代谢物质,它在我们的身体内扮演着促进脂肪酸燃烧、提供能量的重要角色。
游离肉碱偏低可能会引起一系列的健康问题,因此我们需要重视游离肉碱的指标检测,并及时做出调整。
2.游离肉碱偏低可能是由于一些慢性疾病、饮食习惯不良、药物影响等引起的。
当我们发现游离肉碱偏低时,要及时就医,排除其他潜在的健康问题,并根据医生的建议进行相应的治疗。
3.我们在日常生活中也可以通过合理饮食、适量运动等方式来预防游离肉碱偏低的发生,保持身体健康。
三、谱系1.谱系指的是一种基因的传承和演化关系,在人类基因研究领域具有重要的意义。
通过对谱系的研究,我们可以更好地了解人类的起源和演化过程,为人类的健康和疾病研究提供重要的科学依据。
2.谱系研究对于人类疾病的预防、诊断和治疗也具有重要的意义。
通过对不同谱系的特点和遗传信息的研究,我们可以更好地了解不同人裙患病的风险和易感性,为个性化医疗提供科学依据。
3.谱系研究还对于人类的社会和文化发展具有重要的意义。

文/ 孟新科(深圳市第二人民医院急诊科主任医师)
高乳酸血症在临床上并不少见,但相
关病因诊断及治疗却很复杂。
若未能及时
做出正确诊断,往往给治疗带来很大不确
定性结果。
血乳酸水平的高低是患者病情严重程
度的重要标志,而且其能否迅速降至正常
水平(<2mmol/L)也是反映疗效的重要
标准。
其中,脓毒症的相关指南把特定时
高乳酸血症临床评估路径
1.评估组织低灌注和恢复再灌注能力
◆休克(分布性、心源性、低血容量和
◆先假定存在组织低灌注,直至证明并
◆根据不同休克病因采取不同治疗手段
2.评估局部组织缺血和针对性治疗
◆肠系膜缺血、肢体缺血、烧伤、创伤、筋膜室综合征、坏死性软组织感染
◆药物因素:利奈唑胺、核苷类逆转录。


《神经病学》神经-肌肉接头疾病和肌肉疾病练习题及答案一、选择题【A;型题】,1.重症肌无力禁用的药物是。
A.氢氯唆嗦B.糖皮质激素C.麻黄碱D.环磷酞胺E.氨基糖昔类抗生素2;重症肌无力最常见的受累肌肉是·A.眼外肌B.咽喉肌C.面肌D.四肢近端肌肉E.四肢远端肌肉3.重症肌无力危象与其他危象不易区别时、可采取的最佳方法是」A.肌内注射新斯的明B.肌内注射苯丙酸诺龙C.静脉注射依酚氯钱D.口服澳毗斯的明E.疲劳试验4.重症肌无力患者合并肺部感染时,下列哪种抗生素不宜应用:·A.青霉素B.氯霉素.C.庆大霉素D.头抱氨节E.甲硝哇月5.由于自身抗体作用子突触前膜电压依赖性钙离子通道使ACh释放减少所致的肌无力是A. Lambert-Eaton syndromeB.美洲箭毒中毒C.肉毒毒素中毒D.重症肌无力E.多发性肌炎6.重症肌无力造成无力的原因是:A.产生AChR抗体使AChR受损或减少B.使胆碱醋酶活性受到抑制导致ACh作用过度延长C.使ACh合成和释放减少D.阻碍钙离子进人神经末梢E.阻碍ACh与AChR结合7. Lambert-Eaton syndrome造成肌无力的原因是A.产生AChR抗体使AChR受损或减少B.使胆碱醋酶活洲性受到抑制导致ACh作用过度延长C.使ACh合成和释放减少D.阻碍钙离子进入神经末梢E.阻碍ACh与AChR结合8.下列哪些不是重症肌无力的特点A.起病隐袭,眼外肌常常首先受累B. 10岁以下儿童眼外肌受累首发者更加常见C.受累肌肉呈病态疲劳性,晨轻暮重D.肌无力分布符合单一神经或神经根受累模式E.平滑肌和括约肌通常不受累9.改善重症肌无力症状的药物是A.糖皮质激素B.免疫抑制剂C.胆碱醋酶抑制剂D.免疫球蛋白E.抗生素10.重症肌无力患者服用胆碱醋酶抑制剂出现不良反应时可给予A.新斯的明B.依酚氯钱C.阿托品D.毛果云香碱E.地西浮11.重症肌无力患者出现危象的主要表现是A.眼睑下垂加重B.吞咽困难加重C.构音障碍加重D.呼吸肌无力E.四肢无力加重12.重症肌无力危象抢救的关键是A.积极控制感染B.肌内注射新斯的明C.给予免疫抑制剂D.辅助通气E.去除使病情加重的诱因13.重症肌无力患者最常伴有A.胸腺瘤B.肺癌C.甲状腺功能亢进D.系统性红斑狼疮E.胸腺增生14.重症肌无力患者眼症状中不包括A.眼睑下垂B.复视C.瞳孔散大,对光反射丧失D._眼球固定E.外展麻痹15.关于周期性瘫痪的描述下列哪项不正确A.我国少见B.男性多见C.本病发作与甲亢的严重程度无关D.发作多在早晨醒来丫运动或饱餐后E.心律失常多见、16.下列哪种肌病有明显的压痛A.进行性肌营养不良症.B.线粒体肌病几C.先天性肌强直D.强直性肌营养不良症E.多发性肌炎17.关于Duchenne型肌营养不良症,下列哪项描述不正确A.为最常见的类型B.主要影响男性,X连锁的隐性遗传病-C.有明显的地理或种族差异D.女性为基因携带者E. 1/3的患儿是MM基因新突变所致18.多发性肌炎的首选治疗是A.糖皮质激素B.免疫抑制剂C.血浆置换D.免疫球蛋白E.高蛋白饮食19.关于Becker型肌营养不良症,下列哪项描述不正确·「A.较少见B. X连锁的隐性遗传C.发病和死亡年龄较晚D.病情进展快E.预后较好20.与进行性肌营养不良发病相关的蛋白是A.抗肌萎缩蛋白B.肌球蛋白C.肌动蛋白D.肌钙蛋白E.原肌球蛋白21.常见的伴有排肠肌假肥大的进行性肌营养不良症的为A. Duchenne型:肌营养不良和肢带型肌营养不良B. Becker型肌营养不良和眼肌型肌营养不良C. Duchenne型肌营养不良和远端型肌营养不良D. Becker型肌营养不良和Duchenne型肌营养不良E. Duchenne型肌营养不良和眼肌型肌营养不良22.进行性肌营养不良症的下列哪项描述不正确A.是一组遗传性肌肉变性病B.病情进展缓慢,症状进行性加重C.累及肢体和头面部肌肉,少数有心肌受累D.对称性肌无力、肌萎缩,可有肌肥大E.肌电图运动单位电位时限增宽、波幅增高,多相波增多23.低钾型周期性瘫痪的常见诱因是A.饱餐B.闷热C.出汗D.大笑E:嗜盐24.低钾型周期性瘫痪属于A.骨骼肌钠离子通道病B.骨骼肌钾离子通道病C.骨骼肌氯离子通道病D.骨骼肌钙离子通道病E.骨骼肌钾离子和氯离子通道病25.周期性瘫痪最常累及的肌肉是A.眼外肌B.咽喉肌C.四肢近端肌肉;D.尿道和肛门括约肌E.肋间肌和月扇肌26.可以出现肌强直的周期性瘫痪是A.低钾型周期性瘫痪B.高钾型周期性瘫痪C.正常血钾型周期性瘫痪D.心律失常型周期性瘫痪E.甲亢性周期性瘫痪27.关于多发性肌炎下列哪项不正确A.四肢近端对称性肌无力B.首发症状多为站立、上下楼和梳头困难C.常伴有肌肉酸痛和压痛D.血沉和血清肌酶正常E.可有颈部肌肉无力,表现为抬头困难28.眼肌型肌营养不良与重症肌无力的主要鉴别依据是A.肌电图B.血清肌酶C.四肢近端肌无力且感觉无异常D.肌肉活检E.家族史29.下列哪项属于骨骼肌氯离子通道病A.进行性肌营养不良症-B.多发性肌炎C.高钾型周期性瘫痪D.低钾型周期性瘫痪E.先天性肌强直症30.进行性肌营养不良症中哪种的病情最严重A.先天型B. Duchenne型C. Becker型D.肢带型E.面肩肪型31.最常见的常染色体显性遗传的肌营养不良类型是A. Duchenne型肌营养不良B. Becker型肌营养不良C.肢带型肌营养不良D.面肩脑型肌营养不良E.眼肌型肌营养不良32. 40岁以上发病的皮肌炎患者须高度警惕A.恶性肿瘤B.系统性红斑狼疮C.类风湿性关节炎D.心脏病E.糖尿病33.多发性肌炎一般不累及A.四肢近端肌肉B.颈部肌肉C.咽喉肌D.呼吸肌E.眼外肌34.下列疾病中属于X性连锁隐性遗传的为A.低钾型周期性瘫痪B.少年近端型脊髓性肌萎缩症C. Andersen syndromeD.先天性肌强直E:假肥大型肌营养不良35.以反复发作的突发性骨骼肌瘫痪为特征的疾病为A.进行性肌营养不良症B.多发性肌炎C.低钾型周期性瘫痪D.少年近端型脊髓性肌萎缩症E.先天性肌强直36.低钾型周期性瘫痪患者24小时口服补钾的总量为A. 5gB. l0gC. 15gD. 20gE. 25g37.患者躯干和四肢近端肌肉叩击后出现局部肌球现象,数秒钟后消失,该体征可见于,A.进行性肌营养不良B.强直性肌营养不良C.多发性肌炎D.低钾型周期性瘫痪E.脊髓性肌萎缩症38.在线粒体病的描述中不正确的是A.是一组由线粒体DNA或核DNA缺陷导致线粒体结构和功能障碍,ATP合成不足所致的多系统疾病B.其共同特征为轻度活动后即感到极度疲乏无力,林息后好转C.肌肉活检可见破碎红纤维二且如病变以侵犯骨骼肌为主,则称为线粒体肌病.E.线粒体病是常染色体显性遗传39.线粒体脑肌病伴高乳酸血症和卒中样发作(MELAS)综合征的临床特点中不包括一A.卒中样发作伴偏瘫、偏盲或皮质盲B.偏头痛C.神经性耳聋D.脑脊液蛋白水平增高E.头颅CT和MRI显示主要为枕叶脑软化,病灶范围与主要脑血管分布不一致,可见基底节钙化40.慢性进行性眼外肌瘫痪无需与下列哪种疾病鉴别A.重症肌无力一B.眼咽型肌营养不良C. Miller Fisher syndromeD.多发性肌炎E.眼肌型肌营养不良41.线粒体肌病和线粒体脑肌病的治疗中不包括A.高蛋白、高碳水化合物和低脂饮食B.静脉滴注ATP及辅酶AC.口服辅酶Q10D.口服左卡尼汀E.免疫抑制剂42. Duchenne型肌营养不良与Becker型肌营养不良共有的临床特点中不包括A. X性连锁隐性遗传B.用卜肠肌假肥大C.远端肢体无力D.血清CK水平增高E.肌电图和肌肉病理呈肌病改变43. Duchenne型肌营养不良症患者不包括下列哪些临床表现.A.男性,12岁以后发病B.鸭步C.Gower征D.排肠肌假肥大E.翼状肩胛44.Duchenne型、肌营养不良患者多在25-30岁死于A.脑庙B.心力衰竭C.呼吸衰竭D.肾功能衰竭E.消耗性疾病45.下列疾病中不属于自身免疫性疾病的有A.重症肌无力B. Lambert-Eaton syndromeC.进行性肌营养不良D.慢性炎症性脱髓鞘性多神经病E.多发性肌炎46.关于进行性肌营养不良症的下列描述哪些不正确A.肌无力和肌萎缩多为对称性B. Duchenne型肌营养不良是抗肌萎缩蛋白基因缺陷所致C. Duchenne型肌营养不良患儿心肌受累较多见D. Becker型肌营养不良的症状与Duchenne型肌营养不良相似,但病情轻微E.眼咽型可见特殊的肌病面容“斧头脸”47.下列哪些疾病属于钠离子通道病A.高钾型周期性瘫痪B.低钾型周期性瘫痪C.强直性肌营养不良D.多发性肌炎E.先天性肌强直48.重症肌无力受累部位是A.前角细胞B.神经肌肉接头C.感觉神经节D.肌肉E.神经末梢49. Duchenne型肌营养不良患者的假性肥大常见于哪些部位A.不会发生B.仅限于肩月甲带肌C.仅限于骨盆带肌D.用腓肠肌E.仅限于大腿肌肉【A2型题】1.男患,20岁,早晨醒来发现四肢无力,不能起床,查体发现四肢肌张力减低,腿反射减低,感觉正常,病理征(一),心电图示U波,最可能的诊断是A.低钾型周期性瘫痪B.正常血钾型周期性瘫痪C.高钾型周期性瘫痪D.多发性肌炎E.疮症性瘫痪2女患,16岁,全身无力1年余,已经诊断重症肌无力,服用嗅毗斯的明后症状减轻。
HHH综合征(高鸟氨酸血症-高氨血症-同型瓜氨酸尿症)概述高鸟氨酸血症-高氨血症-同型瓜氨酸尿症(Hyperornithinaemia-hyperammonaemia-homocitrullinuria Syndrome,HHHS)是由位于13q14染色体上编码线粒体鸟氨酸转运蛋白的SLC25A15基因突变引起的一种常染色体隐性遗传病。
因鸟氨酸转移蛋白1(ornithine transporter1,ORNT1)缺乏,导致尿素循环功能障碍。
HHHS是一种具有高度临床变异性的异质性疾病,轻型临床表现为学习困难和轻微神经系统受累症状,重型表现为昏迷、嗜睡、肝脏体征和癫痫发作。
新生儿期发病的患者具有严重的临床表现,除此之外,没有证据表明发病年龄与疾病严重程度之间存在直接关系。
病因和流行病学鸟氨酸是一种碱性氨基酸,参与尿素循环并发挥重要作用。
ORNT1蛋白位于线粒体膜上,可将鸟氨酸从细胞质转运到线粒体内参与尿素循环。
编码ORTN1的SLC25A15基因发生突变使其功能发生缺陷,引起以下生化反应:①线粒体内鸟氨酸含量下降,血中鸟氨酸含量升高;②鸟氨酸不能与氨甲酰磷酸充分反应,引起氨甲酰磷酸堆积;③累积的氨甲酰磷酸通过旁路代谢生成乳清酸,也可以与赖氨酸结合生成同型瓜氨酸,引起同型瓜氨酸增多:④尿素循环受阻后导致游离氨蓄积,形成高氨血症,引发相应的临床表现。
HHHS的发病率极低,全世界至今仅报道了100多例患者,男女比例为2∶1。
欧美HHHS发病率约为1/350000,目前国内缺乏相应的流行病学资料,但有病例报道。
临床表现HHHS临床症状、体征主要由高氨血症及高鸟氨酸血症和高同型瓜氨酸尿症引起,以神经系统症状为主,受ORNT1缺陷程度影响,临床表现严重程度亦不同。
发病年龄可以从新生儿期到成年期(晚发型),并且具有广泛的表型谱。
1.新生儿期约12%的患者在新生儿期发病。
出生后的24~48小时内无明显症状,随后出现与高氨血症相关的症状[包括嗜睡、困倦、拒食、呕吐,伴呼吸性碱中毒和(或)癫痫发作]。
线粒体脑肌病伴高乳酸血症和脑卒中样发作综合征的临床、影像学及病理学特点谢成娟;汪凯【摘要】目的探讨线粒体脑肌病伴高乳酸血症和脑卒中样发作(MELAS)综合征的临床、影像及病理学特点.方法回顾性分析7例MELAS综合征患者的临床资料.结果本组患者中,男1例,女6例,平均年龄21.3岁;均为卒中样起病伴癫痫发作,其中视力减退6例,运动后乏力5例,认知功能障碍2例,精神异常1例.头颅MRI检查示顶、枕叶和(或)颞叶T1WI/T2 WI长或稍长信号及Flair高信号病灶,呈脑回样改变,均未见强化.4例患者肌肉活检发现有肌纤维变性,横纹消失或断裂,肌膜下出现不规则的不整红边纤维(RRF).2例行基因检测发现有mtDNA A3243G位点突变.结论MELAS综合征好发于青少年,以卒中样起病伴癫痫发作,可有视听力减退以及认知功能障碍等;影像学特征为病变主要累及双侧大脑半球后部皮质,呈脑回样改变;肌肉活检发现RRF.【期刊名称】《临床神经病学杂志》【年(卷),期】2014(027)001【总页数】3页(P5-7)【关键词】线粒体脑肌病伴高乳酸血症和脑卒中样发作综合征;临床特点;影像学;病理学【作者】谢成娟;汪凯【作者单位】230022,安徽医科大学第一附属医院神经内科;230022,安徽医科大学第一附属医院神经内科【正文语种】中文【中图分类】R746线粒体脑肌病是由于线粒体DNA(mtDNA)或核DNA(少数)发生缺失或突变,线粒体结构或功能异常导致的以脑和肌肉受累为主的多系统疾病,属于母系遗传病[1,2]。
1984年 Pavlakis 等[3]首次报道了线粒体脑肌病伴高乳酸血症和脑卒中样发作(MELAS)综合征以后,临床医生对MELAS综合征的认识和研究不断提高。
但由于其临床表现多样,容易误诊漏诊,导致预后较差。
如何早诊断、早治疗一直是临床实践中的难题。
本研究对7例MELAS综合征患者的临床资料进行回顾性总结,以探讨其临床、影像及病理学特点。