分子几何构型优化的初步比较
- 格式:doc
- 大小:1.29 MB
- 文档页数:6

化学分子的几何结构分析化学分子是由多个原子通过化学键相互连接而成的,它们中包含着复杂的几何结构,几何结构的形状和类型决定了分子的性质和反应。
化学分子的几何结构分析是分析分子结构和反应机理的重要手段。
本文将探讨化学分子的几何结构分析方法和应用。
一、分子结构分子结构是分子内成键原子的相对位置。
在分子中,原子间的成键通过原子之间的电子对共享来形成。
成键原子所构成的点称作原子核间的成键域。
原子核间的成键域可以是单个成键对,也可以是多个成键对的集合以及孤对电子。
分子结构的三维空间位置和取向可以通过分子轨道理论来解释。
在分子轨道理论中,分子中的原子轨道相互超出,因此电子云的体积将分成不同的分子轨道。
这些分子轨道可以是电子互相远离的反键绑定轨道,也可以是电子相互靠近的成键轨道。
分子轨道的能量不同,取决于原子成键的方向和成键时电子的位置。
分子结构的分析需要研究这些分子轨道的构成和相互关系。
二、分子几何形状分子几何形状是由原子间成键的角度和键长所确定的空间结构。
分子几何形状的描述通常采用分子几何构型。
常见的分子几何构型包括线性、三角形、四面体、平面三角形、八面体和三角双锥等。
由于化学键是有方向性的,分子几何构型对于反应和性质的影响是决定性的。
分子几何构型的确定需要采用不同的实验和理论方法。
三、实验方法实验方法是确定分子几何构型最常用的手段。
实验方法主要包括X射线结晶学、光电子能谱学和微波光谱学三种。
X射线结晶学是最常见的分析物质结构的方法之一。
它利用物质对X射线的散射实现分子结构分析。
利用X射线晶体学技术,可以获得物质的结构信息,包括原子间距离、键角等。
光电子能谱学是发现有机化学物质和无机材料结构的重要手段之一。
它是通过将样品表面吸引出电子并测量其能量来分析物质的电子结构及其物化性质。
微波光谱学技术是利用微波辐射的吸收和发射来研究分子内的状态和转化。
此方法仅适用于具有偶极矩的分子,通常用于确定分子的旋转和振动状态和分子几何构型。

分子结构模型的构建及优化计算分子结构模型的构建是化学研究和计算化学领域的重要一环,对于理解分子的性质和行为具有重要意义。
优化计算则是对构建的分子结构模型进行调整和优化,以求得最稳定和最符合实验结果的结构体系。
本文将介绍分子结构模型的构建方法以及常用的分子结构优化计算方法。
一、分子结构模型的构建1.实验室试验方法:实验室试验方法通过实验手段确定分子的构型和结构。
常用的实验方法包括谱学方法(如红外光谱、拉曼光谱、核磁共振等)、X射线方法和电子显微镜等。
这些实验方法可以提供分子的一些基本信息,例如键长、键角、晶胞参数等。
不过该方法需要实验设备和实验条件,有时也受到实验技术的限制。
2. 理论计算方法:理论计算方法主要通过量子力学计算、分子力学模拟和分子动力学模拟等,从基本粒子的角度计算分子的结构和性质。
在量子力学计算中,常用的方法有Hartree-Fock(HF)方法、密度泛函理论(DFT)方法、紧束缚模型(TB)方法等。
在分子力学模拟和分子动力学模拟中,常用的方法有分子力学(MM)方法、分子动力学(MD)方法等。
二、分子结构优化计算分子结构优化计算是对构建的分子结构模型进行调整和优化的过程,以找到最稳定和最符合实验结果的结构体系。
1.线性规划方法:线性规划方法是寻找一个解向量,使得目标函数最小或最大。
在分子结构优化计算中,可以通过线性规划方法来优化分子结构的内部参数,如键长、键角等。
2. Monte Carlo方法:Monte Carlo方法是一种通过随机抽样的方式来进行优化计算的方法。
在分子结构优化计算中,Monte Carlo方法可以通过随机调整分子的内部参数,以整个构象空间,寻找最稳定的构象。
3.遗传算法:遗传算法是通过模拟生物进化过程来进行优化计算的方法。
在分子结构优化计算中,可以将每一个分子结构看作一个个体,通过交叉、变异等操作模拟自然选择,以寻找最优解。
4.分子动力学模拟:分子动力学模拟是通过求解分子的运动方程,模拟分子的运动和变化过程。

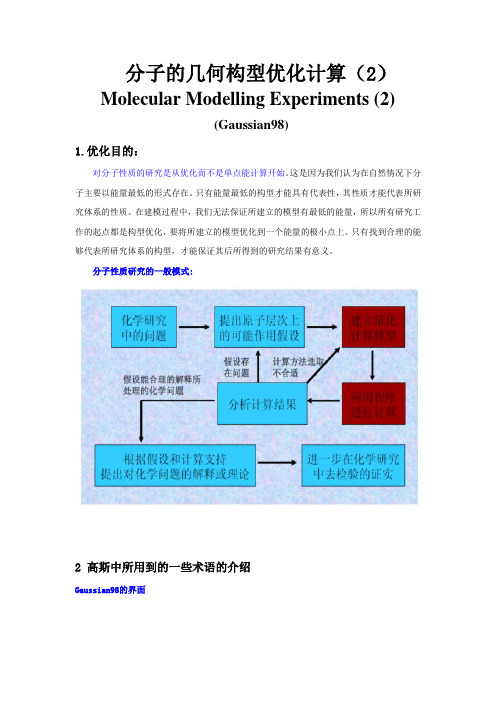
分子的几何构型优化计算(2)m ents (2)a n98)1.优化目的:对分子性质的研究是从优化而不是单点能计算开始。
这是因为我们认为在自然情况下分子主要以能量最低的形式存在。
只有能量最低的构型才能具有代表性,其性质才能代表所研究体系的性质。
在建模过程中,我们无法保证所建立的模型有最低的能量,所以所有研究工作的起点都是构型优化,要将所建立的模型优化到一个能量的极小点上。
只有找到合理的能够代表所研究体系的构型,才能保证其后所得到的研究结果有意义。
分子性质研究的一般模式:2 高斯中所用到的一些术语的介绍a n98的界面2.1势能面在不分解的前提下,分子可以有很多个可能的构型,每个构型都有一个能量值,所有这些可能的结构所对应的能量值的图形表示就是一个势能面,势能面描述的是分子结构和其能量之间的关系,以能量和坐标作图。
根据分子中的原子数和相互作用形式,有可能是二维的,也有可能是多维的。
势能面上的每一个点对应一个具有一个能量的结构。
能量最低的点叫全局最小点,局域最小点是在势能面上某一区域内能量最小的点,一般对应着可能存在的异构体。
鞍点是势能面上在一个方向有极大值而在其他方向上有极小值的点,通常对应的都是过渡态。
优化的目的就是找到势能面上的最小点,因为这个点所对应的构型能量最低,是最稳定的。
2.2确定能量最小值构型优化就是找体系的最小点或鞍点。
能量的一阶导(也就是梯度,注意在数学中,一阶导表示着函数的变化趋势,一阶导为零就表明找到了极值点,这是确定最小值的数学基础)是零,这表明在这个点上的力也是零(因为梯度的负值是力)。
我们把势能面上这样的点称为静态点(也就是上面所说的极小点)。
所有成功的优化都会找到一个静态点,虽然有时找到的静态点并不是想要的静态点。
程序从输入的分子构型开始沿势能面进行优化计算,其目的是要找到一个梯度为零的点。
计算过程中,程序根据上一个点的能量和梯度来确定下一步计算的方向和步幅。
梯度其实就是我们所说的斜率,表示从当前点开始能量下降最快的方向。

分子的立体构型及杂化方式的判断方法1.VSEPR理论:VSEPR理论是根据分子中的电子对的排斥效应来预测分子的立体构型。
根据这一理论,分子中的电子对(包括原子间键合电子对和孤对电子对)会尽量远离彼此,从而使得分子呈现出一定的几何形状。
通过确定分子中原子的中心原子、其他原子和孤对电子对的数量,可以用VSEPR理论预测分子的构型。
2.分子对称性:分子的对称性可以提供有关分子构型的重要信息。
通过观察分子的对称轴、平面反转中心、镜面反射面等,可以推断分子的几何结构。
例如,如果一个分子具有旋转轴,那么该分子很可能是线性的;若分子具有反射面,则可能是平面的。
3. 分子轨道理论:分子轨道理论描述了分子中电子的分布情况,指出原子轨道会经过一个或多个杂化过程而形成分子轨道。
通过观察分子中的化学键及相关杂化轨道的形状,可以推断出分子的杂化方式。
例如,如果分子中存在 sigma 键,则可判断该分子中原子的杂化方式为 sp 杂化;如果分子中存在 pi 键,则可判断杂化方式为 sp2 或 sp3 杂化。
4.光谱学方法:光谱学方法可以通过分析分子在不同波长下吸收和发射的电磁辐射来研究分子的构型和杂化。
例如,红外光谱可以提供分子中不同键弯曲、拉伸等振动模式的信息,从而帮助确定分子的立体构型;核磁共振光谱可以提供分子中不同核磁共振信号的信息,从而揭示出分子中不同原子的环境和构型。
5.计算化学方法:计算化学方法可以通过理论计算来预测分子的构型和杂化。
例如,量子力学方法(如密度泛函理论)可以计算分子的电子结构和能量,从而推测分子的构型和杂化方式。
总之,分子的立体构型和杂化方式可以通过VSEPR理论、分子对称性、分子轨道理论、光谱学方法和计算化学方法等多种途径来判断。
这些方法的使用取决于具体分子的性质和研究目的。

分子结构的秘密从简单到复杂分子结构是化学和生物科学中一个至关重要的概念,它不仅是理解物质性质的基础,也为各种科学研究提供了理论支持。
分子是由原子通过化学键结合而成的,而分子结构则指的是这些原子如何在三维空间中排列。
分子结构可以分为简单和复杂两种类型,随着科学的发展,我们对分子结构的理解已经从最初的简单模型深入到复杂的立体结构。
本文将逐步揭示分子结构的秘密,从基础的分子概念到复杂的生物分子的结构解析。
分子的基本概念首先,必须明确“分子”这一概念。
分子是由两个或多个原子通过共价键结合形成的最小化合物单位。
这意味着一个分子的组成和结构将直接影响其性质,例如熔点、沸点、溶解度等。
根据不同的原子组合,分子可以是同种元素构成(如氢气H₂、氧气O₂等),也可以是不同元素构成(如水H₂O、二氧化碳CO₂等)。
原子的组成与排列原子的定义: 原子是物质的基本单位,由质子、中子和电子组成。
质子的数量决定了元素的性质,而电子的排列则影响原子的反应性。
键的形成: 原子间通过化学键结合形成分子,主要包括三种类型:共价键、离子键和金属键。
在共价键中,原子通过共享电子来结合;在离子键中,原子通过电荷吸引力结合;金属键则是金属原子共享其外层电子。
分子的表示: 分子的化学式可以用元素符号和数字表示,如H₂O 表示一个水分子,其中两个氢原子和一个氧原子通过共价键结合。
分子的几何结构初步了解了基本概念后,我们来探讨分子的几何结构。
分子的几何形状对其化学性质有着深远影响。
根据VSEPR理论(价层电子对排斥理论),不同数目的电子对会导致不同的几何形状。
例如:线形:当中心原子都有两个取代基时,形成线形结构,如CO₂。
折线型:当中心原子有两个键合基和一个孤立电子对时,如水H₂O。
平面三角形:当中心原子有三个取代基时,如BF₃。
四面体:当中心原子有四个取代基时,如甲烷CH₄。
此外,立体异构现象也与分子的空间构型密切相关,相同化学式但不同空间排列的分子称为立体异构体。
分子空间构型判断方法
分子空间构型判断方法是用来确定分子的立体构型,即分子中各个原子的相对排列方式。
常用的分子空间构型判断方法包括:
1. 空间平面判断法:通过判断各个原子的平面关系,来确定分子中的平面构型,如平面刚性构型判断法和平面柔性构型判断法。
2. 键长角度判断法:通过测量化学键的长度和键角的大小,来确定分子中的构型,如键长角度分析法和键角关系判断法。
3. 分子力场法:通过构建分子力场模型,计算分子在不同构型下的能量差异,来确定分子的最稳定构型,如分子力场方法和分子动力学模拟方法。
4. 图解法:通过绘制分子的扭转曲线,来确定分子的扭转构型,如扭转角度和扭转能量图解法。
5. 分子轨道法:通过计算分子的分子轨道能级和电子密度分布,来确定分子的构型,如分子轨道法和密度泛函理论方法。
以上方法根据不同的分子特性和目标可以灵活组合使用,以得到更准确的分子空间构型判断结果。
Chem 3D分子构型该怎么优化很多的化学领域的专业人士都知道,分子的性质往往是有分子的结构所决定的,但是在实验室研究的过程中很难观察到稳定分子的结构,这往往是因为中间体寿命过短或者是混合物难以分离造成的,这个时候就需要通过计算化学来预测,即构型优化。
下面来给大家分享一下使用Chem 3D化学绘图软件轻松优化分子构型的两种方法。
所有计算化学研究分子性质均是从优化分子结构开始,在自然情况下分子主要以能量最低的子女格式存在,所以低能的分子结构具有代表性,这样也才能保证得到的计算结构有意义,Chem 3D软件使用MM2分子力学和Gamess量子两种方法来优化构型。
1、MM2分子力学优构型优化方法使用Chem 3D软件MM2分子力学优化构型的操作步骤是绘制出化学结构之后,依次选择Calculations/MM2/Minimize Energy(最小化化学能)命令,如下图所示:MM2分子力学优构型优化方法接着会弹出Minimize Energy对话框,“Display Every nth Iter用于显示每轮迭代信息”、“Copy Measurements to Output Bo用于控制输出每轮结构参数”、“Minimum RMS是构型收敛标准”。
另外注意一下,MM2分子力学方法计算量小,适合于大体系有机分子的构型优化。
2、Gamess量子化学软件包构型优化方法Gamess量子化学软件包进行构型优化的原理是Chem 3D根据初始分子模型计算能量和梯度,然后决定写一部结构调整的方向的步长,根据各原子受力情况和位移大小判断是否收敛,若没有则继续重复上面的过程直到力和位移的变化均达到收敛标准。
在Chem 3D软件中的操作方法是:绘制出分子结构之后,依次点击Calculations/GAMESS Interface/Minimize,随后会弹出如下图所示的GAMESS对话框。
Gamess量子化学软件包构型优化方法对话框Job&Thery选项卡的Method选项可以更改计算方法,Basis Set 是基组类型,Wave Function是波函数类型,通过Polarization、Diffuse和Exponent可以添加或弥散基函数,Opt.Algorithm用于修改构型优化方法,最后的两个选项Spin Multiplicity和Net Charge指的是体系电荷和自选多重度。
判断分子空间几何构型的简单方法
分子空间几何的构型是研究分子的化学性质和关联性质的重要参考,由此可以知道物质的密度、电荷密度分布特征和电子激发态的迁移以及分子的位移和变形,从而能够更好的预测分子的后续反应特性。
在实际的研究过程中,如何判断分子空间几何构型,是一个关于分子结构与功能之间关系的重要步骤。
最常用的方法是通过实验观察,采用X射线衍射或NMR技术,对分子中的原子进行测看和定位,获取分子空间几何构型的具体信息,从而判断该物质的构型模式。
但是,随着科技的发展,计算机模拟技术也在不断完善,它将实验所需的大量重复工作,转化为电脑的模拟运算,从而使得对分子构型的判断非常简单。
另一种常用的方法是对分子的构型进行理论推演,根据分子中所包含的原子及其电子表象力、共价键角以及化学键长等特征,以某种特定斥力场下的极限决定方程来预测分子的几何构型、结构和性质,从而进行判断。
而这种方法的优势在于可以较为直观的得到分子的轨道构型,而对于复杂的分子构型来说也是一种很直接的判断方法。
综上所述,判断分子空间几何构型的简单方法有实验观察和计算理论推演两种。
实验观察可以采用X射线衍射或NMR技术,而理论推演则是根据分子中所包含的原子及其相关特征,利用某种特定斥力场下的极限决定方程来判断分子的构型。
虽然这两种方法都能够较快得到预期结果,但在实际中,如何根据判断出来的结果应用到研究中还需要仔细结合该分子特性深入思考。
分子几何构型优化的初步比较
摘要对《CRC物理与化学手册》[1]第78版中收集的已知实验构型的多原子分子NH3, 在HF、MP2的级别上进行了构型优化的初步比较.优化采用基组STO-3G、6-31G(d、p) 6-311G(d、p), 以及6-311G(2d、p). 对同一方法比较了基组函数, 对同一基组函数则比较了HF 和MP2 方法.结果表明,键长的平均绝对偏差(单位:pm)由大到小的顺序为: MP2/STO-3G, HF/STO-3G, HF/6-311G(d、p),HF/6-31G(d、p),HF/6-311G(2d、p), MP2/ 6-31G(d、p) ,MP2/6-311G(d、p).
引言量子化学优化分子几何构型的理论和应用日趋成熟和程序化[2-4],使研究新型配
合物的物质结构,配合物中间体,反应机理研究的手段向前迈进了大大的一步.可供选择的分子几何构型优化的方法很多,但何种理论方法较为精确,一直是相关研究工作者关心和期望得到明确答案的问题.在一些很高级别的能量计算和发展精确模型化学计算方法中[ 5],这一问题更加突出.可以认为分子几何构型优化的可靠性是一切精确计算的基本保证.由于理论上对于分子几何构型优化尚无规律和系统方法克可循目前只能通过一些系统性的比较来探讨这个问题.对于一些有机小分子构型优化的比较,已有文献报道,但仍不够充分,而对于化学建类型变化较多的无机分子的系统比较则较少.[5 ,6]. 构型优化的计算量往往较大,因此寻找和事先确定适当的构型优化方法,已成为计算化学领域中一个共同感兴趣的问题.本文就是从此问题出发, 以NH3分子构型的优化为基点, 以STO-3G、6-31G(d、p), 6-311G(d、p), 以及6-311G(2d、p)为基组,在HF 和MP2级别上进行全面的理论计算和比较.由于计算数据较多,会引起篇幅较大,本文将不一一列出数据,而是直接报导计算和分析统计结果.
1. 实验数据
不同的实验方法得到的数据有所不同,且实验中有平衡构型和各种振动态(多为基态及低激发振动态)的平动构型r0以及同位素平均构型r e等,鉴于低振动态势阱的非谐性对结构参数的影响一般不大,本文将不区分r e和r0等,而全部按文献[1]数据实录.
NH3 为C3v对称点群,:,为了计算输入的方便, 将NH3分子构型中的一些实验参数作了等价转化.
2. 计算方法
分子几何构型优化理论方法建立在对分子势能面认识的基础上,其物理模型和数学模型是十分清晰的.这里不再赘述.本文中,我们对NH3分子分成不同基组和不同方法分别讨论.为了突出氢原子基组中加入p极化函数的必要性,重点比较了不同基组态STO-3G和6-31G(d,p), 6-311G(d、p)等.同时对目前普遍采用的HF 方法和MP2 方法进行了同一基组态计算精度的比较分析.,比较其计算度和精度, 分析其偏差原因. .所有计算用Gaussian-98W[2 ] 成. NH3分子的实验键长多数精确到0.01pm,计算结果也精确至0.01pm.
3. 结果与分析
3.1 HF—method下面列出了NH3分子优化前后的结构示意图、计算结果和比较分析表.
所有数据表明,所有理论计算和实验数据値有很好的吻合.最大偏差是HF/STO-3G, 计算表明,它导致优化能量比实验値高.当然,几何构型偏差(AD)的纯数值大小并不能说明它将导致分子能量误差的大小, 因为分子的势阱深度即力常数千差万别. 这里将主要讨论总体的平均绝对偏差(AAD).
NH3分子优化前的构型 RHF-OPT (STO-3G)后的构型
RHF-OPT法NH3分子键长的平均偏差(AD)和平均绝对偏差(AAD)
由键长的基组平均偏差比较可以看出, 6-31G(d,p), 6-311G(2d,p)的优化效果要明显强于另两个基组态.下面再来比较键角和偶极矩的平均差:
通过键角和偶极矩的平均偏差比较,可以看出,相同的HF 方法,在不同的基组上优化的计算精度表现不同.偏差最大的在键长上,键角计算精度很接近,偶极矩的绝对偏差则很大.可能是因为分子空间因素的原因,但其偏差是随不同基组态的选用而逐渐趋向于精确度较高的方向的.综上分析比较,可以近似得出,当选用HF 方法优化分子构型在6-31G(d,p), 6-311G(d,p), 6-311G(2d,p)基组态上操作时,由于考虑了氮(N)原子的d轨道的极化和
氢(H)原子的p轨道极化函数,尽管计算量较大,但优化效果较好.,和实验値十分吻合.
PHF—OPT[6-31G(d,p)]后的构型 RHF—OPT[6-311G(d,p)]后的构型
3.2 MP2-method在STO-3G、6-31G(d、p), 6-311G(d、p)基组态上对NH3分子进行结构优化,由于分子优化后的构型图宏观上没有区别,所以只简单的给出两个基组态6-31G(d,p), 6-311G(d,p)上的优化构型.NH3分子优化后的构型图列于下方
OPT-MP2(STO-3G)优化后的几何构型 OPT-MP2 [6-311G(d,p)]优化后的几何构型
键长和键角,偶极矩的相应偏差计算并分别列在下表中:
同3.1 的分析,可以看到采用MP2方法优化分子几何构型,不同基组仍会使得到的计算精度有很大差异.由键长的平均偏差变化可以看出,是否考虑氢原子和氮原子的外层极化函数p 轨道和d轨道对分子的几何构型优化有很大影响,亦直接影响确定分子的几何结构.相反,这几种基组对键角和偶极矩的影响则很小.可见在优化多原子分子时时一定要尽可能的多考虑到成物原子的外层轨道极化,即在计算时要加入相应的p轨道和d轨道因素,所以如果要选用MP2 优化分子构型时,优先选用或最好选用6-311G(d,p)基组态.
3.3 HF和 MP2 两种方法的比较
通过在STO-3G、6-31G(d、p), 6-311G(d、p)共同基组态上分别采用两种方法即HF和MP2进行NH3分子的结构优化,可以看出不同方法和基组态的选择正如许多文献指出的一样,会导致构型优化的精度有很大差异,这两种方法该如何优先选择呢?在选定方法之后,又如何来确定分子的计算基组态呢?现在尝试通过比较分析,回答问题的答案.两种方法的平均绝对偏差列于下表:
通过比较分析,得知在构型优化时,多原子分子中除氢外的原子的外层极化函数d轨道和氢原子的p轨道必须加入,否则会很大地影响计算结果. 而在考虑了原子外层极化函数的情况下, MP2法则将会使计算结果大大改善,效果明显,所以MP2法要优于HF 法;因此,可以初步得出结论: 要选用STO-3G对分子优化,则要优先选用HF法; 但如果要使用MP2方法做构
型优化,则以6-311G(d、p)基组为最好.但最好选用后者,这样才会得到比较和实验値相吻合的计算数据.
分析比较结果也表明, NH3分子键角的平均偏差都在1.0°和0.4°之内.其中,以(d,2d)
极化函数的基组得到的键角平均绝对偏差为最大,这可能与d 极化函数的方向性较强有关.
.
4. 结论
本文的结果表明,给氢原子增加p极化函数可以较大改善NH3分子即氢化物的键长,建议进行构型优化时最好给氢原子加入p极化函数.MP2方法的优化效果虽较好可计算量较大.MP2/6-31G(d,p) 在6-31G(d,p)的基组上较大地改进了所有分子的构型优化效果.不过, MP2也未能给出优化键长的最好结果.不过,文中所用的两种方法对键角的计算都比较好.在不考虑计算量的情况下,对NH3分子优化后其键长的平均绝对偏差由大到小的顺序排列如下: MP2/STO-3G, HF/STO-3G, HF/6-311G(d、p),HF/6-31G(d、p),HF/6-311G(2d、p), MP2/ 6-31G(d、p) ,MP2/6-311G(d、p).这种情况可以推广到多原子分子.我们在需要进行构型优化时,可以以此来参考, 在忽略计算量的情况下,尽可能的达到较精确的结果.
参考文献:
1. Handbook of chemistry and physics 78t th
2. Gaussian 98,Rev.A.Frish,M J.Trucks G W,schlegel H B, et al .Gaussian Inc,Pittsburgh PA ,1998
3Schlegel H B. Modern Electronic Structure Theory,Ed,Yarkary D R,Singapore: World Scientific Publish,1995.
4Tang Auchin, Li Qianshu. Molecular Reaction Kinetics, Changchun: Jilin University Press,1989.
5. Acta.Phys.Chem.Sin. (物理化学学报), 649 .Vol 16 ,No 7 2000
6. Acta.Phys.Chem.Sin. (物理化学学报), 395 ,V ol 19 , No 4 2002
7.J. C. I . C.(无机化学学报) 394 V ol 18 No 4 2002
8.Chinese .J Stru Chem(结构化学) 214 .V ol 21. No 2 2002
9.Chem .J Chin. Univ (高等学校化学学报) 99~104. Vol.21No.12000.
分子几何构型优化的初步比较------计算化学期终报告
年级2002 级
专业无机化学
姓名刘三会
指导教师缪强
二O O 三年元月八日。