分子几何构型判断3
- 格式:ppt
- 大小:734.00 KB
- 文档页数:69

高中杂化轨道判断技巧杂化轨道是化学中的一个重要概念,它描述了原子中电子云的一种混合状态。
在高中化学中,学生需要掌握杂化轨道的概念,并能够判断分子中原子的杂化轨道类型。
本文将介绍几种高中杂化轨道判断技巧,帮助学生更好地掌握这个概念。
一、根据分子几何构型判断杂化轨道类型分子几何构型是指分子中原子的空间排列方式。
在杂化轨道理论中,不同的分子几何构型对应着不同的杂化轨道类型。
因此,通过观察分子几何构型,我们可以初步推断原子的杂化轨道类型。
例如,当分子几何构型为线性时,我们可以判断其中原子的杂化轨道为sp;当分子几何构型为三角形平面时,我们可以判断其中原子的杂化轨道为sp2;当分子几何构型为四面体时,我们可以判断其中原子的杂化轨道为sp3。
二、根据分子中原子的电子数判断杂化轨道类型在分子中,原子的杂化轨道类型还可以通过观察其电子数来判断。
通常情况下,一个原子的杂化轨道类型取决于其原子轨道中存在的电子数。
例如,当一个原子的s轨道和两个p轨道中共有四个电子时,我们可以判断其杂化轨道类型为sp3。
当一个原子的s轨道和一个p轨道中共有两个电子时,我们可以判断其杂化轨道类型为sp。
三、根据分子中原子的化学键数判断杂化轨道类型在分子中,原子的杂化轨道类型还可以通过观察其与其他原子形成的化学键数来判断。
通常情况下,一个原子的杂化轨道类型取决于其与其他原子形成的化学键数。
例如,当一个原子与三个其他原子形成共价键时,我们可以判断其杂化轨道类型为sp3。
当一个原子与两个其他原子形成共价键时,我们可以判断其杂化轨道类型为sp2。
四、根据分子的反应活性判断杂化轨道类型在分子中,原子的杂化轨道类型还可以通过观察其反应活性来判断。
通常情况下,原子的杂化轨道类型与其反应活性有关。
例如,当一个原子的杂化轨道为sp3时,其反应活性通常较低。
而当一个原子的杂化轨道为sp时,其反应活性通常较高。
总结杂化轨道是化学中的一个重要概念,学生需要掌握其概念和判断方法。

分子几何构型
分子几何构型是指分子中原子在空间的相对排列关系。
不同的分子几何构型对应于不同的化学键和键长、键角等参数。
了解分子几何构型对于了解物质的性能与其内部结构的关系具有十分重要的意义。
常见的分子几何构型包括直线型、平面正三角形、正四面体、三角锥形、折线形、平面形、折面形、平面正四边形、正四面梯形等。
这些构型可以通过实验测定的键长、键角等数据来确定。
在理论化学中,可以使用理论方法预测分子的几何构型。
例如,价键理论(VB理论)可以用线性变分法处理H₂分子,得到三个等同的sp2杂化轨道,形成平面正三角形的构型。
此外,分子轨道理论也可以用来预测分子的几何构型。
总之,分子几何构型是化学键和分子结构研究的重要内容之一,对于理解物质的性质和反应机理具有重要意义。


分子或离子空间构型的判断方法
1.空间构型的可解析法:根据分子或离子的几何构型和化学键的长度、角度以及存在的非共面情况,可以通过几何推算来判断其空间构型。
例如
在VSEPR理论中,根据中心原子周围键的种类和数量以及孤对电子的影响,可以预测分子的空间构型。
2.实验技术法:通过实验手段来确定分子或离子的空间构型。
例如,
X射线晶体学可以通过测定晶体中原子的位置来确定其空间构型。
3.分子模拟法:借助于计算机模拟和量子化学计算的手段,可以通过
分子力场、量子力学计算等方法来预测分子或离子的空间构型。
这种方法
可以考虑更多的因素,如电荷分布和随机热运动的影响,从而更准确地判
断空间构型。
4.光谱技术法:在分子或离子存在谐振,激发或振动的情况下,通过
观察其产生的光谱特性,如红外光谱、拉曼光谱等,可以得出分子或离子
的空间构型信息。
以上是几种常用的判断分子或离子空间构型的方法,不同的方法在实
际应用中有其适用范围和局限性。
对于一些简单的分子和离子,几何推算
方法通常可以给出较为准确的空间构型信息。
而对于复杂的大分子和离子,分子模拟和实验技术往往是更可靠和准确的方法。
通过综合运用这些方法,可以对分子和离子的空间构型进行全面的判断和研究。
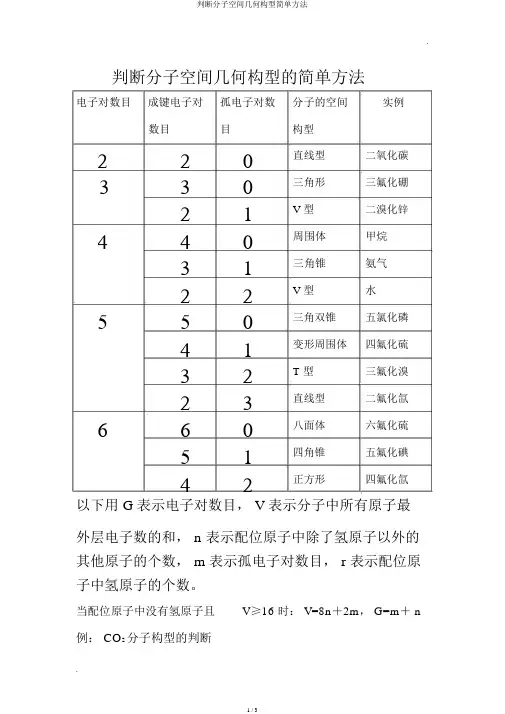
判断分子空间几何构型的简单方法电子对数目成键电子对孤电子对数分子的空间实例数目目构型220 33021 4403122 550413223 6605142直线型二氧化碳三角形三氟化硼V 型二溴化锌周围体甲烷三角锥氨气V 型水三角双锥五氯化磷变形周围体四氟化硫T 型三氟化溴直线型二氟化氙八面体六氟化硫四角锥五氟化碘正方形四氟化氙以下用 G表示电子对数目, V 表示分子中所有原子最外层电子数的和, n 表示配位原子中除了氢原子以外的其他原子的个数, m表示孤电子对数目, r 表示配位原子中氢原子的个数。
当配位原子中没有氢原子且V≥16 时: V=8n+2m, G=m+ n 例: CO2分子构型的判断V=4+ 6× 2=8n+ 2m,这里 n=2,∴ m=0,∴ G=m+ n=0+2=2,所以 CO2的分子构型为直线型BF3分子构型的判断V=3+ 7× 3=8n+ 2m,这里 n=3,∴ m=0,∴ G=m+ n=0+3=3,所以 BF3的分子构型为三角形PCl5分子构型的判断V=5+ 7× 5=8n+ 2m,这里 n=5,∴ m=0,∴ G=m+ n=0+5=5,所以 PCl5的分子构型为三角双锥SF4分子构型的判断V=6+ 7× 4=8n+ 2m,这里 n=4,∴ m=1,∴G=m+ n=1+4=5,所以 SF4的分子构型为变形周围体BrF3分子构型的判断V=7+ 7× 3=8n+ 2m,这里 n=3,∴ m=2,∴G=m+ n=2+3=5,所以 BrF3的分子构型为 T 型SF6分子构型的判断V=6+ 7× 6=8n+ 2m,这里 n=6,∴ m=0,∴ G=m+ n=0+6=6,所以 SF6的分子构型为八面体XeF4分子构型的判断V=8+ 7× 4=8n+ 2m,这里 n=4,∴ m=2,∴G=m+ n=2+4=6,所以 XeF4的分子构型为正方形当配位原子中有氢原子且 V< 16 时: V=2r + 8n + 2m,G=m+ n+ r 。

价电子对互斥理论(VSEPR )与分子几何构型(原载《结构化学问题选讲》,杨宗璐等编,科学出版社2000年)多原子分子的结构比双原子分子要复杂得多,因此一般需要用一组键长和键角的数据来表征其几何构型.原则上可以通过越来越多的计算方案来求解波动方程,得到所需的结合能、平衡几何构型和电荷密度.另一方面,针对大量的复杂分子,人们总是力图采用简化的模型和图象来推理和总结多原子分子中电子行为的规律.价电子对互斥理论(VSEPR )正是这样一种简明、直观、可广泛预测许多简单多原子分子结构的一种方法.价电子对互斥理论(VSEPR )实际上是建立在定域模型基础上的静电理论.按照定域键模型,分子中的电子可划分为:内层电子、孤对电子及成键电子(价电子)等独立组份.分子的能量则与这些独立组份能量及它们的相互作用有关.通过核间距的调整使键长和键角的相互适应,产生了能量最低的几何结构.此外,价电子的分布又受到Pauli 不相容原理和静电库仑作用的支配,除了要求分子中的电子尽量配对外,为使体系电子云分布具有能量最低值,还要求已配对的电子之间彼此尽可能远离,以降低库仑排斥作用.以ALmEn 代表多原子分子,其中A 为中心原子,L 为配位原子,E 则为A 原子价层上的孤对电子对,m 为价层总电子对数P,则可借上述价电子互斥理论提出判断分子几何构型的以下规则:n +1.一定数目的价电子对必定与一定的几何构型相联系.假定价电子对的中心与原子A的距离都是R,为使彼此间斥力最小,价电子对等距离地排布在以R为半径的球面上,形成规则的多面体.根据立体几何的知识,当P为2时,呈直线形(键角为180o);P为3 时呈平面三角形(键角为120o);P为4时呈四面体形(键角为10928′o );P为5时呈三角双锥形;P为6时呈八面体形;P为7 时则呈五角双锥形等.2. 价电子对互斥理论将分子的共价双键或三重键均视为一个电子对来计算.由于多重键中的电子对数多,占据的空间比一个单键电子对大,它对其它单键电子对的排斥作用较强,使分子中含多重键的键角变大,不同键电子对之间排斥作用的大小顺序为三键排斥>双键排斥>单键排斥显然,多重键的存在会影响分子的几何构型.例如 X 2C=0 型分子属于三角形构型,各键角标准值应为120o,不含双键的键角受到前者的影响应小于120o .具体实例见表1.表1 键角的比较X2C=0 ∠XCO ∠XCXF2C=0 123.20112.50Cl2C=0 124.30111.30H2C=0 1210118.00(NH2)2 C =0 1210118.00X2C=CX2∠XCX ∠XCCH2C=CH2116.60121.70(CH3)2C=CH2109012503.用于推测分子结构时,不能忽略孤对电子和成键电子的区别.因为孤对电子对比键电子对更集中于原子核附近,因而增加了排斥能.此外,成键电子对于两个原子核的吸引,电子云显得不如孤对电子的“肥大”,对邻近电子对的斥力也小.孤对电子与键对电子间的排斥强度符合以下排序:孤对/孤对>孤对/键对>键对/键对.一般情况下,由于孤对与键对的不同取向可形成一组异构构型,以上的排斥作用顺序可以帮助辨认能量最低的稳定构型.当某个构型中孤对与孤对的交角小于或等于90o,则必定是不稳定构型.例如,ICl4-的6对价电子对指向正八面体6个顶点,由于只有二对孤对电子,故可形成两种不同的异构构型(见图1).在平面正方形的构型中,两对孤对电子相距最远,应为稳定构型.又如,在SF4,ClF3和IF2-的价电子层中,价电子对数P均为5,但孤对电子对数分别为1、2、3.根据价电子对互斥理论,孤对电子对应选择赤道方向的位置,这样可使与孤对电子对互成90o相互作用的键对数目最少.从图2可知,在它们各自的最稳定构型中,孤对电子对均处于三角平面内.4. 配位原子的电负性增大或中心原子电负性减小,也会使键角偏离标准值.端原子电负性增加时,共享电子对将偏向配位体,从而减少成键电子对之间的斥力,键角的度数随之减小.例如OF2的键角(103.2o)比OH2的键角(104.5o)要小.NF3的键角(102o)比NH3的键角(107.3o)也要小.此外,中心原子的半径增大使与端原子之间的键长增加,减小键电子对之间的排斥力,使键角缩小.实例参见表2.表2 键角的比较分子式 键角 分子式 键角NH3107.30H2O 104.50PH393.30H2S 92.20AsH391.80H2Se 91.00SbH391.30H2Te 89.005.价电子对的相邻电子对越多,所受斥力就越大,距离原子核也越远.当配位数为5和7时,分子呈三角双锥和五角双锥构型,这时主轴上两对电子受到的斥力较大,因而键长较长.例如PCl5分子中,垂直方向的键长为219pm,水平方向键长为204pm,见图3.价电子对互斥理论能够广泛地用于定性预测各类ABn型分子的几何构型,解释键长和键角变化,偏离标准值的规律性,但也有少数化合物的推测出现例外.例如BaI2,SrCl2都是弯曲形构型而非预计的直线性型.此外,对过渡金属化合物几何构型的判断也有一定的局限性.实际上,应用价电子对互斥理论的分子体系,都有一个共同的特点,即分子中所有电子都是成对的.如果分子中有未成对的电子,则不能简单地以此方法来推测分子的几何构型.对于不具有半满或全满d轨道的过渡元素,由于d轨道上电子分布不对称,将使分子形状不规则化,无法用价电子对互斥理论作出判断,因而配位场理论则能说明得更好.价电子对互斥理论被看成是杂化轨道理论简化了的方法.而它相当于只考虑了中心原子利用杂化轨道成键后,各电子对之间的相互排斥,因而只能作出大致的定性说明.而杂化轨道理论则还考虑了整个的杂化过程,并可得出定量的结果,如计算键角等.。





判断分子空间几何构型的简单方法以下用G表示电子对数目,V表示分子中所有原子最外层电子数的与,n表示配位原子中除了氢原子以外的其它原子的个数,m表示孤电子对数目,r表示配位原子中氢原子的个数。
当配位原子中没有氢原子且V≥16时:V=8n+2m,G=m+n 例:CO2分子构型的判断V=4+6×2=8n+2m,这里n=2,∴m=0,∴G=m+n=0+2=2,所以CO2的分子构型为直线型BF3分子构型的判断V=3+7×3=8n+2m,这里n=3,∴m=0,∴G=m+n=0+3=3,所以BF3的分子构型为三角形PCl5分子构型的判断V=5+7×5=8n+2m,这里n=5,∴m=0,∴G=m+n=0+5=5,所以PCl5的分子构型为三角双锥SF4分子构型的判断V=6+7×4=8n+2m,这里n=4,∴m=1,∴G=m+n=1+4=5,所以SF4的分子构型为变形四面体BrF3分子构型的判断V=7+7×3=8n+2m,这里n=3,∴m=2,∴G=m+n=2+3=5,所以BrF3的分子构型为T型SF6分子构型的判断V=6+7×6=8n+2m,这里n=6,∴m=0,∴G=m+n=0+6=6,所以SF6的分子构型为八面体XeF4分子构型的判断V=8+7×4=8n+2m,这里n=4,∴m=2,∴G=m+n=2+4=6,所以XeF4的分子构型为正方形当配位原子中有氢原子且V<16时:V=2r+8n+2m, G=m+n+r。
例:CH4分子构型的判断V=4+1×4=2r+8n+2m,这里r=4,n=0,∴m=0, ∴G=m+n+r=0+0+4=4,所以CH4的分子构型为四面体NH3分子构型的判断V=5+1×3=2r+8n+2m,这里r=3,n=0,∴m=1,∴G=m+n+r=1+0+3=4,所以NH3的分子构型为三角锥 H2O分子构型的判断V=6+1×2=2r+8n+2m,这里r=2,n=0,∴m=2,∴G=m+n+r=2+0+2=4,所以H2O的分子构型为V型HClO分子构型的判断V=1+7+6=2r+8n+2m,这里r=1,n=1,∴m=2,∴G=m+n+r=2+1+1=4,所以HClO的分子构型为V型HNO分子构型的判断V=1+5+6=2r+8n+2m,这里r=1,n=1,∴m=1,∴G=m+n+r=1+1+1=3,所以HNO的分子构型为V型HCN分子构型的判断V=1+4+5=2r+8n+2m,这里r=1,n=1,∴m=0,∴G=m+n+r=0+1+1=2,所以HCN的分子构型为直线型注:以上所介绍的方法只有当V为偶数时才适用,当V为奇数时,只需将V与G瞧成比它们大1的偶数即可!。
知识探究: 化学键和分子结构Chemical Bonds and Structures of Molecules这一章是化学的核心,因为结构决定性质。
如白磷、红磷的结构不同,性质也不同;石墨、金刚石和C 60等的结构不同,性质也不同。
这一章重点讲授共价键、离子键、金属键以及分子几何构型、金属晶体、原子晶体和离子晶体的晶体结构。
另外我们也讨论分子间的作用力以及对分子晶体的一些性质的影响。
共价键与分子几何构型Covalent Bonds and Molecular Geometric Structure*(要求了解)一、经典共价键理论(Classical Covalent Bond Theory ) − LewisStructure (Octet Rule)(八电子规则)1.基本思想:当n s 、n p 原子轨道充满电子,会成为八电子构型,该电子构型是稳定的,所以在共价分子中,每个原子都希望成为八电子构型(H 原子为2电子构型)。
2.共价分子中成键数和孤电子对数的计算:例如:P 4S 3、HN 3、N +5、H 2CN 2(重氮甲烷)、NO -3 (1) 计算步骤:a .令n o − 共价分子中,所有原子形成八电子构型(H 为2电子构型)所需要的电子总数b .令n v − 共价分子中,所有原子的价电子数总和c .令n s − 共价分子中,所有原子之间共享电子总数 n s = n o - n v ,n s /2 = (n o - n v ) / 2 = 成键电子对数d .令n l − 共价分子中,存在的孤电子数。
(或称未成键电子数)3.Lewis 结构式的书写P 4S 3HN 3HN N N H N N HNNNN 5+,,,N N NNNNNN NNNNNN N N NCH 2N 2(重氮甲烷) ,HCHNN HC HN N当Lewis 结构式不只一种形式时,如何来判断这些Lewis 结构式的稳定性呢?如HN 3可以写出三种可能的Lewis 结构式,5N +可以写出四种可能的Lewis 结构式,而重氮甲烷只能写出两种可能的Lewis 结构式。
判断分子空间几何构型的简单方法以下用G表示电子对数目,V表示分子中所有原子最外层电子数的和,n表示配位原子中除了氢原子以外的其它原子的个数,m表示孤电子对数目,r表示配位原子中氢原子的个数。
当配位原子中没有氢原子且V≥16时:V=8n+2m,G=m+n 例:CO2分子构型的判断V=4+6×2=8n+2m,这里n=2,∴m=0,∴G=m+n=0+2=2,所以CO2的分子构型为直线型BF3分子构型的判断V=3+7×3=8n+2m,这里n=3,∴m=0,∴G=m+n=0+3=3,所以BF3的分子构型为三角形PCl5分子构型的判断V=5+7×5=8n+2m,这里n=5,∴m=0,∴G=m+n=0+5=5,所以PCl5的分子构型为三角双锥SF4分子构型的判断V=6+7×4=8n+2m,这里n=4,∴m=1,∴G=m+n=1+4=5,所以SF4的分子构型为变形四面体BrF3分子构型的判断V=7+7×3=8n+2m,这里n=3,∴m=2,∴G=m+n=2+3=5,所以BrF3的分子构型为T型SF6分子构型的判断V=6+7×6=8n+2m,这里n=6,∴m=0,∴G=m+n=0+6=6,所以SF6的分子构型为八面体XeF4分子构型的判断V=8+7×4=8n+2m,这里n=4,∴m=2,∴G=m+n=2+4=6,所以XeF4的分子构型为正方形当配位原子中有氢原子且V<16时:V=2r+8n+2m, G=m+n+r。
例:CH4分子构型的判断V=4+1×4=2r+8n+2m,这里r=4,n=0,∴m=0,∴G=m+n+r=0+0+4=4,所以CH4的分子构型为四面体NH3分子构型的判断V=5+1×3=2r+8n+2m,这里r=3,n=0,∴m=1,∴G=m+n+r=1+0+3=4,所以NH3的分子构型为三角锥 H2O分子构型的判断V=6+1×2=2r+8n+2m,这里r=2,n=0,∴m=2,∴G=m+n+r=2+0+2=4,所以H2O的分子构型为V型HClO分子构型的判断V=1+7+6=2r+8n+2m,这里r=1,n=1,∴m=2,∴G=m+n+r=2+1+1=4,所以HClO的分子构型为V型HNO分子构型的判断V=1+5+6=2r+8n+2m,这里r=1,n=1,∴m=1,∴G=m+n+r=1+1+1=3,所以HNO的分子构型为V型HCN分子构型的判断V=1+4+5=2r+8n+2m,这里r=1,n=1,∴m=0,∴G=m+n+r=0+1+1=2,所以HCN的分子构型为直线型注:以上所介绍的方法只有当V为偶数时才适用,当V 为奇数时,只需将V和G看成比它们大1的偶数即可!。
分子几何构型公式
分子几何构型公式是一种描述分子三维结构的表示方法。
它使用原子之间的连接关系以及电子对的排布来确定分子的形状。
以下是一些常见的分子几何构型及其相应的公式:
1. 线性(Linear):M-A-B,其中 M 为中心原子,A 和 B 是两个连接在 M 上的原子。
例子:CO₂
2. 角形(Trigonal Planar):M-A-B-C,其中 M 为中心原子,
A、B、C 是连接在 M 上的原子。
例子:BF₃
3. 四面体(Tetrahedral):M-A-B-C-D,其中 M 为中心原子,
A、B、C、D 是连接在 M 上的原子。
例子:CH₄
4. 三角双锥(Trigonal Bipyramidal):M-A-B-C-D-E,其中 M 为中心原子,A、B、C、D、E 是连接在 M 上的原子。
例子:PCl₅
5. 八面体(Octahedral):M-A-B-C-D-E-F,其中 M 为中心原子,A、B、C、D、E、F 是连接在 M 上的原子。
例子:SF₆
这些是常见的几种分子几何构型,每种构型都有对应的原子连接关系和电子排布方式。
请注意,这些公式表示的是分子的理想化结构,实际分子的构型可能受到原子间的电子云排斥、键角扭曲等因素的影响而发生变化。
判断分子空间几何构型的简单方法
分子空间几何的构型是研究分子的化学性质和关联性质的重要参考,由此可以知道物质的密度、电荷密度分布特征和电子激发态的迁移以及分子的位移和变形,从而能够更好的预测分子的后续反应特性。
在实际的研究过程中,如何判断分子空间几何构型,是一个关于分子结构与功能之间关系的重要步骤。
最常用的方法是通过实验观察,采用X射线衍射或NMR技术,对分子中的原子进行测看和定位,获取分子空间几何构型的具体信息,从而判断该物质的构型模式。
但是,随着科技的发展,计算机模拟技术也在不断完善,它将实验所需的大量重复工作,转化为电脑的模拟运算,从而使得对分子构型的判断非常简单。
另一种常用的方法是对分子的构型进行理论推演,根据分子中所包含的原子及其电子表象力、共价键角以及化学键长等特征,以某种特定斥力场下的极限决定方程来预测分子的几何构型、结构和性质,从而进行判断。
而这种方法的优势在于可以较为直观的得到分子的轨道构型,而对于复杂的分子构型来说也是一种很直接的判断方法。
综上所述,判断分子空间几何构型的简单方法有实验观察和计算理论推演两种。
实验观察可以采用X射线衍射或NMR技术,而理论推演则是根据分子中所包含的原子及其相关特征,利用某种特定斥力场下的极限决定方程来判断分子的构型。
虽然这两种方法都能够较快得到预期结果,但在实际中,如何根据判断出来的结果应用到研究中还需要仔细结合该分子特性深入思考。
if3中i的几何构型在化学中,分子的几何构型对于分子的性质和反应具有重要影响。
if3是一种分子式为IF3的化合物,其中包含一个氟原子和三个碘原子。
本文将探讨if3中i的几何构型及其相关性质。
if3分子的结构if3是一种由一个氟原子和三个碘原子组成的分子。
根据VSEPR理论(Valence Shell Electron Pair Repulsion,价壳层电子对斥力理论),分子中的电子对会互相排斥,力求使其间距最大化。
根据这一理论,我们可以推测if3分子的几何构型。
VSEPR理论VSEPR理论基于以下假设:1.原子之间和孤立电子对之间的排斥力相等。
2.电荷密度越高,排斥力越强。
3.具有相同数目的电荷密度(双键或孤立电子对)之间的排斥力相等。
根据这些假设,我们可以预测if3分子中i原子周围电荷密度最低,并且与其他原子之间存在最小排斥力。
if3中i原子的几何构型根据上述推理,我们可以得出if3分子中i原子的几何构型为三角双锥形。
在这种构型中,i原子位于一个平面上,三个碘原子则位于该平面之外。
if3分子的性质if3分子的几何构型对于其性质具有重要影响。
下面将介绍if3分子的一些性质。
极性由于if3分子中包含一个氟原子和三个碘原子,而氟原子比碘原子更电负,因此if3分子是极性分子。
在三角双锥形几何构型中,氟原子位于平面上方,而碘原子则位于平面之外。
这种不均匀的电荷分布导致了分子整体呈现极性。
反应性if3分子由于其极性和几何构型的特殊性质,在化学反应中表现出一定的特异性。
例如,在与其他化合物发生反应时,if3可以作为路易斯酸(电荷亲合剂)与路易斯碱(电荷供体)发生相互作用。
此外,由于if3分子中i原子周围存在较高的电荷密度,它也可能参与其他相互作用或反应。
用途if3分子在化学领域具有一定的用途。
例如,它可用作催化剂,在某些有机合成反应中发挥重要作用。
此外,if3也可用于其他研究领域,如材料科学和表面化学等。