局部给药制剂微生物限度检查
- 格式:ppt
- 大小:1.10 MB
- 文档页数:15
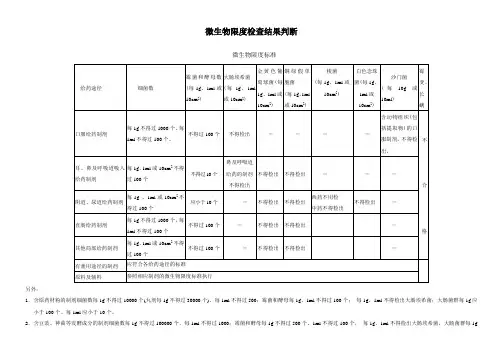
微生物限度检查结果判断
微生物限度标准
另外:
1.含原药材粉的制剂细菌数每1g不得过10000个(丸剂每1g不得过30000个)、每1ml不得过500;霉菌和酵母每1g、1ml不得过100个;每1g、1ml不得检出大肠埃希菌;大肠菌群每1g应小于100个、每1ml应小于10个。
2.含豆豉、神曲等发酵成分的制剂细菌数每1g不得过100000个、每1ml不得过1000;霉菌和酵母每1g不得过500个、1ml不得过100个,每1g、1ml不得检出大肠埃希菌,大肠菌群每1g
应小于100个、每1ml应小于10个。
3.用于表皮或黏膜不完整的含原药粉的局部给药制剂:细菌数每1g或10cm2不得过1000个、每1ml不得过100个;霉菌和酵母每1g、1ml或10cm2不得过100个;每1g、1ml不得检出金黄色葡萄球菌、铜绿假单孢菌。
4.用于表皮或黏膜完整的含原药粉的局部给药制剂:细菌数每1g或10cm2不得过10000个、每1ml不得过100个;霉菌和酵母每1g、1ml或10cm2不得过100个;每1g、1ml不得检出金黄色葡萄球菌、铜绿假单孢菌。

二、微生物限度标准(2010版药典二部附录P115,一部P88)(一)致病菌•口服制剂每g或每ml不得检出大肠埃希菌,含动物药及脏器的药品同时不得检出沙门菌•含中药原生药粉还不得检出大肠菌群(大肠杆菌)•外用每g或每ml不得检出铜绿假单胞菌(绿脓杆菌)、金黄色葡萄球菌;梭菌;大肠埃希菌(眼、鼻、呼吸)•阴道、创伤、溃疡用制剂同时不得检出破伤风杆菌。
一次为准,检出以不合格处理(二)活螨不得检出(三)细菌与霉菌1、要求无菌或标示无菌的制剂:应符合无菌检查规定3、局部给药制剂1)用于手术、烧伤或严重创伤的:应符合无菌检查规定2)用于表皮或粘膜不完整、含药材原粉的:•细菌数:≤1000 cfu /g或10cm2,≤100 cfu /ml•霉菌和酵母菌数:≤100 cfu /g、ml或10cm2•金黄色葡萄球菌、铜绿假单胞菌:不得检出/g、ml或10cm2 (各外用制剂均同)3)用于表皮或粘膜完整、含药材原粉的:细菌数:≤10 000 cfu /g或10cm2,≤100 cfu/ml霉菌和酵母菌数:≤100 cfu /g、ml或10cm2金黄色葡萄球菌、铜绿假单胞菌:不得检出/g、ml或10cm2 (各外用制剂均同)4)眼部给药制剂已全提升为无菌制剂原:细菌数:≤10 cfu /g或ml霉菌和酵母菌数:每1g或1ml不得检出金黄色葡萄球菌、铜绿假单胞菌、大肠埃希菌:每1g或1ml不得检出5)耳、鼻及呼吸道给药制剂细菌数:≤100 cfu /g、ml或10cm2霉菌和酵母菌数:≤10 cfu /g、ml或10cm2金黄色葡萄球菌、铜绿假单胞菌:不得检出/g、ml或10cm2大肠埃希菌:鼻及呼吸道给药制剂, 不得检出/ g、ml或10cm26)阴道、尿道给药制剂细菌数:≤100 cfu /g或ml霉菌和酵母菌数:≤10 cfu /g或ml金黄色葡萄球菌、铜绿假单胞菌、白色念珠菌:不得检出/g、ml或10cm27)直肠给药制剂细菌数:≤1000 cfu /g,≤100 cfu /ml霉菌和酵母菌数:≤100 cfu /g或ml金黄色葡萄球菌、铜绿假单胞菌:不得检出/g或ml8)其它局部给药制剂细菌数:≤100 cfu /g、ml或10 cm2霉菌和酵母菌数:≤100 cfu/g、ml或10cm2金黄色葡萄球菌、铜绿假单胞菌:不得检出/g、ml或10cm24、含动物组织及动物类原药材粉(蜂蜜、王浆、动物角、阿胶除外)的口服制剂:每10g或10ml还不得检出沙门氏菌5、有兼用途径制剂:应符合合途径的标准6、霉变、长螨者:以不合格论。

微生物限度检查标准操作规程一、引言。
微生物限度检查是指在制药生产过程中,对原料药、辅料、中间体、制剂等药品进行微生物检查的一项重要工作。
微生物限度检查的目的是为了保证药品的质量和安全,防止因微生物污染而引起的药品变质和危害患者健康的风险。
本操作规程的目的是规范微生物限度检查的操作流程,确保检查结果准确可靠。
二、适用范围。
本操作规程适用于制药企业进行微生物限度检查的操作人员,包括检验员、操作人员等。
三、术语和定义。
1. 微生物限度,指药品中允许存在的微生物的最大数量,通常以菌落形成单位(CFU)或细菌总数来表示。
2. 微生物限度试验,是指对药品中的微生物进行检查的一种实验方法,包括细菌总数、大肠杆菌、铜绿假单胞菌、霉菌和酵母菌等指标。
3. 标准菌株,指已经鉴定并保存在实验室中的具有代表性的微生物菌株。
四、操作流程。
1. 样品准备。
(1)取样品,按照规定的取样方法,从所检查的药品中取得代表性样品。
(2)样品处理,根据检测要求,对样品进行必要的处理,如稀释、均质等。
2. 培养基制备。
(1)准备培养基,根据检测要求,准备所需的培养基,包括琼脂培养基、液体培养基等。
(2)灭菌处理,对培养基进行高压蒸汽灭菌处理,确保培养基的无菌状态。
3. 菌种接种。
(1)接种方法,根据检测要求,选择适当的接种方法,包括平板法、涂布法等。
(2)接种数量,根据微生物限度试验的要求,按照规定的接种数量接种标准菌株。
4. 培养条件。
(1)温度控制,根据不同的微生物菌种,控制培养的温度,通常为30-35摄氏度。
(2)培养时间,根据微生物限度试验的要求,培养一定的时间,通常为24-48小时。
5. 菌落计数。
(1)观察菌落,根据培养基上的菌落形成情况,进行菌落计数。
(2)结果判定,根据菌落计数的结果,判定样品中微生物的数量是否符合规定的限度要求。
五、质量控制。
1. 内部质量控制,每批次微生物限度试验前,进行内部质量控制,确保实验条件的稳定性和可靠性。

微生物限度检查标准操作规程目的:建立微生物限度检查标准操作规程。
范围:适用于微生物限度检查法的操作。
职责:QC检验人员对本标准实施负责。
执行标准:《中国药典》2020年版四部140页。
规程:1. 简述1.1微生物计数法系用于能在有氧条件下生长的嗜温细菌和真菌的计数。
1.2当本法用于检查非无菌制剂及其原、辅料等是否符合规定的微生物限度标准时,应按下述规定进行检验,包括样品的取样量和结果的判断等。
除另有规定外,本法不适用于活菌制剂的检查。
1.3本检查法可采用替代的微生物检查法,包括自动检测方法,但必须证明替代方法等效于药典规定的检查方法。
1.4微生物计数试验应在受控洁净环境下的局部洁净度不低于B级的洁净空气区域内进行。
检验全过程必须严格遵守无菌操作,防止再污染,防止污染的措施不得影响供试品中微生物的检出。
洁净空气区域、工作台面及环境应定期进行监测。
1.5如供试品有抗菌活性,应尽可能去除或中和。
供试品检查时,若使用了中和剂或灭活剂,应确认其有效性及对微生物无毒性。
供试液制备时如果使用了表面活性剂,应确认其对微生物无毒性以及与所使用中和剂或灭活剂的相容性。
2 计数方法2.1计数方法包括平皿法、薄膜过滤法和最可能数法(简称MPN法)。
MPN法用于微生物计数时精确度较差,但对于某些微生物污染量很小的供试品,MPN法可能是更适合的方法。
供试品检查时, 应根据供试品理化特性和微生物限度标准等因素选择计数方法,所选的方法必须具备检测充足样品量的能力,以保证所获得的试验结果能够判断供试品是否符合规定。
所选方法的适用性须经确认。
2.2计数培养基适用性检查和供试品计数方法适用性试验供试品微生物计数中所使用的培养基应进行适用性检查。
2.3供试品的微生物计数方法应进行方法适用性试验,以确认所采用的方法适合于该产品的微生物计数。
2.4若检验程序或产品发生变化可能影响检验结果时,计数方法应重新进行适用性试验。
注:当需用玫瑰红钠琼脂培养基测定霉菌和酵母菌总数时,应进行培养基适用性检查,检查方法同沙氏葡萄糖琼脂培养基。



微生物限度检查标准操作规程《微生物限度检查标准操作规程》一、目的微生物限度检查是药品、食品、化妆品等产品质量的重要指标之一,其目的是为了保障产品的安全性和稳定性。
本标准操作规程旨在规范微生物限度检查的操作流程,确保检查结果的准确性和可靠性。
二、范围本标准操作规程适用于药品、食品、化妆品等产品的微生物限度检查,包括大肠菌群、金黄色葡萄球菌、霉菌和酵母菌等微生物指标的检查。
三、检查设备和试剂1. 培养基:根据不同的微生物指标选择适当的培养基,如大肠培养基、Mannitol盐琼脂、Sabouraud葡萄糖琼脂等。
2. 培养皿:使用符合规定的试验培养皿。
3. 培养箱:保证培养条件的恒温培养箱。
4. 其他常用实验用具:包括无菌采样容器、吸管、移液器、无菌培养皿等。
四、操作流程1. 样品采集:从产品中采集适量样品,保持无菌状态。
2. 制备稀释液:根据样品的特性,选择适当的稀释液将样品进行适当稀释。
3. 接种培养:将稀释后的样品接种在适当的培养基上,并在符合条件的培养箱中进行培养。
4. 观察和统计:观察培养皿中微生物的生长情况,统计出微生物的数量。
5. 结果判定:根据标准规定的微生物限度标准,判断检测结果是否符合规定。
五、质量控制1. 实验员必须进行严格的无菌操作训练,并使用正确的个人防护用具。
2. 培养基的质量必须符合规定,避免培养基带有细菌污染。
3. 检查设备必须经过定期的检查和校准,保持正常的工作状态。
4. 标本的采集、处理、保存和运送必须符合规定,避免外界的污染和干扰。
六、结果报告根据检测结果和规定的限度标准,出具检测报告,标明产品的微生物限度是否符合规定,以及具体的检测结果数据。
七、总结微生物限度检查是产品质量检验的重要环节,严格按照标准操作规程进行操作,可以保证检测结果的准确性和可靠性。
实验员在操作过程中也需严格遵守规程,做好个人防护措施,确保实验室的安全和卫生。
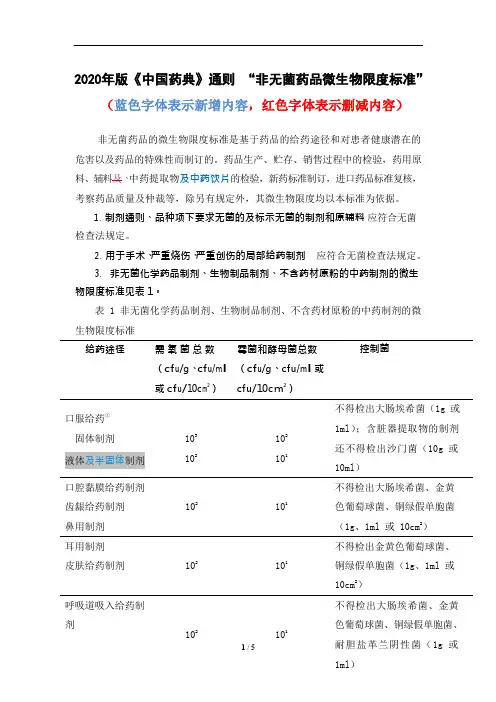
2020年版《中国药典》通则“非无菌药品微生物限度标准”(蓝色字体表示新增内容,红色字体表示删减内容)非无菌药品的微生物限度标准是基于药品的给药途径和对患者健康潜在的危害以及药品的特殊性而制订的。
药品生产、贮存、销售过程中的检验,药用原料、辅料及、中药提取物及中药饮片的检验,新药标准制订,进口药品标准复核,考察药品质量及仲裁等,除另有规定外,其微生物限度均以本标准为依据。
1.制剂通则、品种项下要求无菌的及标示无菌的制剂和原辅料应符合无菌检查法规定。
2.用于手术、严重烧伤、严重创伤的局部给药制剂应符合无菌检查法规定。
3.非无菌化学药品制剂、生物制品制剂、不含药材原粉的中药制剂的微生物限度标准见表 1。
表 1 非无菌化学药品制剂、生物制品制剂、不含药材原粉的中药制剂的微生物限度标准给药途径需氧菌总数(cf u/g、cf u/m l或cf u/10c m2)霉菌和酵母菌总数(cf u/g、c fu/m l或cfu/10cm2)控制菌口服给药①固体制剂液体及半固体制剂103102102101不得检出大肠埃希菌(1g 或1ml);含脏器提取物的制剂还不得检出沙门菌(10g 或10ml)口腔黏膜给药制剂齿龈给药制剂鼻用制剂102101不得检出大肠埃希菌、金黄色葡萄球菌、铜绿假单胞菌(1g、1ml 或 10cm2)耳用制剂皮肤给药制剂102101不得检出金黄色葡萄球菌、铜绿假单胞菌(1g、1ml 或10cm2)呼吸道吸入给药制剂102101不得检出大肠埃希菌、金黄色葡萄球菌、铜绿假单胞菌、或 10ml ) 阴道、尿道给药制剂102101不得检出金黄色葡萄球菌、铜绿假单胞菌、白色念珠菌(1g 、1ml 或 10cm 2);中药制剂还不得检出梭菌(1g 、 1ml 或 10cm 2)直肠给药 固体制剂103102 不得检出金黄色葡萄球菌、铜绿假单胞菌(1g 或 1ml )其他局部给药制剂102102不得检出金黄色葡萄球菌、铜绿假单胞菌(1g 、1ml 或 10cm 2)注 ①化学药品制剂和生物制品制剂若含有未经提取的动植物来源的成份及矿物质还不得检出沙门菌(10g 或 10ml )。

药品微生物限度标准非无菌药品的微生物限度标准是基于药品的给药途径和对患者健康潜在的危害以及药品的特殊性而制订的。
药品生产、贮存、销售过程中的检验,化学药品原料药、中药提取物及辅料的检验,新药标准制订,进口药品标准复核,考察药品质量及仲裁等,除另有规定外,其微生物限度均以本标准为依据。
非无菌制剂的总需氧菌数、总霉菌及酵母菌数测定照附录×××检查;非无菌制剂的控制菌检查照附录×××检查。
本限度标准解释如下:101CFU:最大可接受限值=20;102CFU:最大可接受限值=200;103CFU:最大可接受限值=2000。
以此类推。
1.制剂通则、品种项下要求无菌的制剂及标示无菌的制剂应符合无菌检查法规定。
2.口服给药制剂2.1 不含药材原粉的口服给药制剂需氧菌总数每1g不得过103cfu。
每1ml不得过102cfu。
霉菌及酵母菌总数每1g不得过102cfu。
每1ml不得过101cfu。
大肠埃希菌每1g或1ml不得检出。
沙门菌含脏器提取物的口服给药制剂每10g或10ml不得检出。
2.2含药材原粉的口服制剂2.2.1不含豆豉、神曲等发酵原粉的口服给药制剂需氧菌总数每1g不得过10000cfu。
每1ml不得过100cfu。
霉菌及酵母菌总数每1g或1ml不得过100cfu。
大肠埃希菌每1g或1ml不得检出。
沙门菌每10g或10ml不得检出。
耐胆盐革兰阴性菌每1g应小于102个。
每1ml应小于101个。
2.2.2 含豆豉、神曲等发酵原粉的口服制剂需氧菌总数每1g不得过100000cfu。
每1ml不得过1000cfu。
霉菌和酵母菌总数每1g不得过500cfu。
每1ml不得过100cfu。
大肠埃希菌每1g或1ml不得检出。
沙门菌每10g或10ml不得检出。
耐胆盐革兰阴性菌每1g应小于102个。
每1ml应小于101个。
3.局部给药制剂3.1 用于手术、烧伤或严重创伤的局部给药制剂应符合无菌检查法规定。

中国药典微生物限度检查法- 概述《中国药典》2010版一部、二部、三部附录的微生物限度检查法内容无差别,故此文方法,以《中国药典》2010版二部附录ⅪJ为例。
微生物限度检查应在环境洁净度10000 级下的局部洁净度100 级的单向流空气区域内进行。
检验全过程必须严格遵守无菌操作,防止再污染。
单向流空气区域、工作台面及环境应定期按《医药工业洁净室(区)悬浮粒子、浮游菌和沉降菌的测试方法》的现行国家标准进行洁净度验证。
供试品检查时,如果使用了表面活性剂、中和剂或灭活剂,应证明其有效性及对微生物无毒性。
除另有规定外,本检查法中细菌及控制菌培养温度为30℃~35℃;霉菌、酵母菌培养温度为23℃~28℃。
检验结果以1g、1ml、10g、10ml、10c㎡为单位报告,特殊品种可以最小包装单位报告。
注:《医药工业洁净室(区)悬浮粒子、浮游菌和沉降菌的测试方法》的现行国家标准分为GB/T16292-2010 医药工业洁净室(区)悬浮粒子的测试方法GB/T16293-2010 医药工业洁净室(区)浮游菌的测试方法GB/T16294-2010 医药工业洁净室(区)沉降菌的测试方法中国药典微生物限度检查法- 检验量和供试液制备检验量检验量即一次试验所用的供试品量(g、ml或cm²)。
除另有规定外,一般供试品的检验量为10g或10ml;膜剂为100cm²;贵重药品、微量包装药品的检验量可以酌减。
要求检查沙门菌的供试品,其检验量应增加20g或20ml(其中10g或10ml用于阳性对照试验)。
检验时,应从2个以上最小包装单位中抽取供试品,膜剂还不得少于4片。
一般应随机抽取不少于检验用星(两个以上最小包装单位)的3倍最供试品。
供试液的制备根据供试品的理化特性与生物学特性,采取适宜的方法制备供试液。
供试液制备若需加温时,应均匀加热,且温度不应超过45℃。
供试液从制备至加入检验用培养基,不得超过1小时。
除另有规定外,常用的供试液制备方法如下。

微生物限度检查法标准操作规程一、目的:建立一个微生物限度检查标准操作规程,规范质检员的操作,保证实验的安全性、准确性。
二、范围:适用于微生物限度检查的产品。
三、责任:QC部质检员对本规程实施负责。
四、内容1.检验依据:《中国药典》2010年版二部。
2. 简述2.1.微生物限度检查法系检查非规定灭菌制剂及其原料、辅料受微生物污染程度的方法。
检查项目包括细菌数、霉菌数、酵母菌数及控制菌数检查。
2.2.微生物限度检查应在环境洁净度10000级下的局部洁净度100级的单向流空气区域内进行。
检验全过程必须严格遵守无菌操作,防止再污染,防止污染的措施不得影响供试品中微生物的检出。
单向流空气区域、工作台面及环境应定期按《医药工业洁净室(区)悬浮粒子、浮游菌和沉降菌的测试方法》的现行国家标准进行洁净度验证。
2.3.供试品检查时,如果使用了表面活性剂、中和剂或灭活剂,应证明其有效性及对微生物无毒性。
2.4.除另有规定外,本检查法中细菌及控制菌培养温度为30~35℃;霉菌、酵母菌培养温度为23~28℃2.5.检验结果以1g、1ml、10g、10ml或10cm2为单位报告,特殊品种可以最小包装单位报告。
3.设备、仪器3.1.设备:3.1.1.洁净实验室:微生物限度检查应有单独的洁净实验室,每个洁净实验室应有独立的净化空气系统。
结构和要求:洁净室应采光良好,避免潮湿、远离厕所及污染区。
操作间与缓冲间应有样品传递窗,出入操作间和缓冲间的门不应直对。
洁净实验室内应六面光滑平整,能耐受清洗消毒。
墙壁与地面、天花板连接处无缝隙,不留死角。
操作间不应安装下水道。
洁净实验室内的照明灯应嵌装在天花板内,室内光照应分布均匀,光照度不低于300LX。
●温度、湿度:洁净实验室内温度应控制在18~26℃,相对湿度最好在40%~60%。
●操作间:操作间应安装空气除菌过滤层流装置。
洁净度不应低于10000级,局部洁净度为100级(或放置同等级净化工作台)。
《中国药典》2010年版二部附录XI J (附录115页)《微生物限度检查法》微生物限度标准非无菌药品的微生物限度标准是基于药品的给药途径及对患者健康潜在的危害而制订的。
药品的生产、贮存、销售过程中的检验,原料及辅料的检验,新药标准制订,进口药品标准复核,考察药品质量及仲裁等,除另有规定外,其微生物限度均以本标准为依据。
1.制剂通则、品种项下要求无菌的制剂及标示无菌的制剂应符合无菌检查法规定。
2.口服给药制剂细菌数每1g不得过l000CFU 。
每lml 不得过100CFU 。
霉菌和酵母菌数每lg或lml 不得过100CFU 。
大肠埃希菌每1g 或lml不得检出.3 .局部给药制剂3.1用于手术、烧伤及严重创伤的局部给药制剂应符合无菌检查法规定。
3.2 耳、鼻及呼吸道吸入给药制剂细菌数每1g、lml 或l0cm2,不得过100CPU 。
霉菌和酵母菌数每1g、lml 或l0cm2,不得过10CPU 。
金黄色葡萄球菌、铜绿假单胞菌每1g、lml 或l0cm2不得检出。
大肠埃希菌鼻及呼吸道给药的制剂,每1g、lml 或l0cm2,不得检出。
3.3 阴道、尿道给药制剂细菌数每1g、lml 或l0cm2,不得过100CFU 。
霉菌数和酵母菌数每1g、lml 或l0cm2应小于10CFU 。
金黄色葡萄球菌、铜绿假单胞菌、白色念珠菌每1g、lml 或l0cm2,不得检出。
3 .4 直肠给药制剂细菌数每1g不得过l000CFU。
每lml 不得过100CFU 。
霉菌和酵母菌数每1g 或lml 不得过100CFU 。
金黄色葡萄球菌、铜绿假单胞菌每lg 或lml 不得检出。
3.5 其他局部给药制剂细菌数每1g、lml 或l0cm2不得过100CFU 。
霉菌和酵母菌数每1g、lml 或l0cm2不得过100CFU 。
金黄色葡萄球菌、铜绿假单胞菌每1g、lml 或l0cm2不得检出。
4.含动物组织(包括提取物)的口服给药制剂每10g 或10ml 还不得检出沙门菌。
氧化锌洗剂微生物限度检查及方法验证实验发表时间:2015-10-28T09:36:57.703Z 来源:《医药前沿》2015年第25期供稿作者:胡泽锴毛腾霄[导读] 成都市食品药品检验研究院氧化锌洗剂为局部给药制剂,本品可采用常规法进行霉菌和酵母菌计数,采用离心沉淀配合薄膜过滤法进行细菌计数和控制菌检查。
胡泽锴毛腾霄(成都市食品药品检验研究院 610000)【摘要】目的:建立氧化锌洗剂的微生物限度检查方法。
方法:采用常规法、薄膜过滤法对氧化锌洗剂的微生物限度检查进行试验。
结果:供试品5种试验菌回收率均>70%,控制菌能正常检出。
结论:应采用薄膜过滤法对细菌数和金黄色葡萄球菌控制菌进行检查;采用常规法对霉菌和酵母菌数以及铜绿假单胞菌控制菌进行检查。
【关键词】氧化锌洗剂;微生物限度检查法;薄膜过滤法【中图分类号】R446 【文献标识码】A 【文章编号】2095-1752(2015)25-0356-03Erification of the Microbial Limit Test for the Zinc Oxide LotionHu Zekai, Mao Tengxiao(Institute for Food and Drug control of Chengdu,Chengdu 610000)【Abstract】 Objective: To establish a method of microbial limit tests for Acne Zinc Oxide lotion. Methods Routine method and membrane filtration method were used for the test of the Zinc Oxide lotion. Results The recoveries were more than 70 % and the quality of control germ check was credible. Conclusion Bacteria and Clostridium sporogenes and Pseudomonas aeruginosa were tested by membrane filtration method.Molds and yeasts were tested by the routine method.【Key words】 Zinc Oxide lotion;Microbial limit tests; Membrane filtration氧化锌洗剂为外用制剂,主要成分为氧化锌、薄荷脑,具有抗菌消炎作用。
微生物限度检查概述及方法第一部分药品微生物限度检查原理;一微生物学基本原理(一)、微生物的概念和类别;所谓微生物(microorgamisms)是一群个体微小结构简单肉眼不能直接看到,必须借助显微镜以及其它手段才能辨别微小生物。
1、结构类型按微生物细胞结构和组成不同可分原核细胞型微生物:代表(细菌)真核细胞型微生物:代表(真菌)非细胞型微生物:代表(病毒)2、营养类型根据微生物的不同营养要求,可分为自养型和异养型两类,又以所利用能源的来源不同,分为化能营养型和光能营养型。
药品中所污染的微生物,从营养型角度看,他大多数异养型,且以腐生性和兼性寄生性化能异养型为主,容易用人工培养繁殖,是药品进行无菌及限度检查的最基本原理。
3、影响微生物生长繁殖的因素1)物理因素:温度、湿度、渗逶压、辐射2)化学因素:营养物质、酸碱度、氧、有毒物质:其影响微生物生长的机制主要有a 使菌体蛋白变性或沉淀b 干扰微生物酶系统各的影响其代谢c 损伤微生物细胞膜,使内容物逸出。
3)生物因素。
在微生物之间,微生物与寄主动植物之间均存在着各种互相作用的关系,主要为寄生、共生、拮抗关系等。
噬菌体与寄主菌、细菌素对某些细菌的抗菌作用,就是生物因素的具体表现。
四、微生物代谢及其在生长检定中的意义微生物生化鉴定试验,所使用的基本原理,就是利用微生物所物有的代谢反应,其中大部分波及到酶的反应1)微生物主要代谢内容A、呼吸作用-根据微生物呼吸作用最终电子受体性质分为呼吸作用,无氧呼吸作用及发酵作用三种,无本质区别,都是物质被氧化,同时另一物质被还原只是物质被氧化分解程度和释放水平不同。
相关的生化鉴反应如过氧化氢酶试验,硝酸盐还原试验,氰化钾试验等。
B、糖的分解有关的生化鉴定反应如糖(醇、苷)类发酵试验,葡萄糖的氧化/发酵试验,甲基试验,甲基红试验,VP试验C、蛋白质和氨基酸的分解相关生化鉴定试验如吲哚试验、明胶液化试验、硫化氢试验等。
(2)、微生物酶酶是由细胞产生的特殊蛋白质或特殊蛋白质复合物,并能催化各种生物化学反应的有机催化剂,与一般无机催化剂相比,酶能在常温压下进行催化,有催化效率高和高度专一性的特点。