实时荧光定量PCR数据分析及常见问题分析
- 格式:ppt
- 大小:2.33 MB
- 文档页数:41

荧光定量 PCR 常见问题分析荧光定量 PCR 常见问题分析 Q1.CT 值出现过晚.1.扩增效率低,反应条件不够优化。
设计更好的引物或探针;改用三步法进行反应;适当降低退火温度;增加镁离子浓度等。
2.PCR 各种反应成分的降解或加样量的不足。
3.PCR 产物太长。
一般采用 80-150bp 的产物长度。
Q2.无 CT 值(信号)出现.信号) 1.反应循环数不够。
一般都要在 35 个循环以上,可根据实验情况增加循环(如至 45cycles),但高于 45 个循环会增加过多的背景信号。
2.检测荧光信号的步骤有误。
一般 SG 法采用72℃延伸时采集,Taqman 法则一般在退火结束时或延伸结束采集信号。
3.引物或探针降解。
可通过 PAGE 电泳检测其完整性。
4.引物或探针的设计,如探针高于引物的温度不够,造成探针未杂交上而产物已延伸的情况。
5.模板量不足。
对未知浓度的样品应从系列稀释样本的最高浓度做起。
6.模板降解。
避免样品制备中杂质的引入及反复冻融的情况。
Q3.标准曲线的线性关系不佳. 1.加样存在误差,使得标准品不呈梯度。
2.标准品出现降解。
应避免标准品反复冻融,或重新制备并稀释标准品。
3.引物或探针不佳。
重新设计更好的引物和探针 4.模板中存在抑制物,或模板浓度过高 Q4.阴性对照也出现明显的起飞. 1.应 mix 或水被污染。
2.引物二聚体的出现。
用 SG 法在 35cycles 以后阴性出现起飞属正常情况,可配合熔解曲线进行分析。
3.反应过程中探针的降解。
用 PAGE 电泳对探针进行检测。
4.如果使用了 ROX 校正,则可能是 ROX 的降解所造成。
Q5.在溶解曲线前是否插入一个同溶解程序初始温度相同的一个保温过程.如果在熔解曲线前没有一个保温过程,则曲线会在前几个循环非常陡峭,使真正的熔解峰型几乎看不到。
Q6.熔解曲线不止一个主峰.1.引物设计不够优化。
应避免引物二聚体和发夹结构的出现。

荧光定量PCR实验数据分析荧光定量PCR(quantitative polymerase chain reaction)是一种基于PCR技术的实验方法,用于定量检测特定DNA序列的丰度。
荧光定量PCR是一种非常重要的分子生物学技术,在基因表达分析、疾病诊断等领域得到广泛应用。
在进行荧光定量PCR实验后,需要对实验数据进行分析,以获取样品中目标DNA序列的丰度信息。
本文将介绍荧光定量PCR实验数据分析的基本步骤和常见方法。
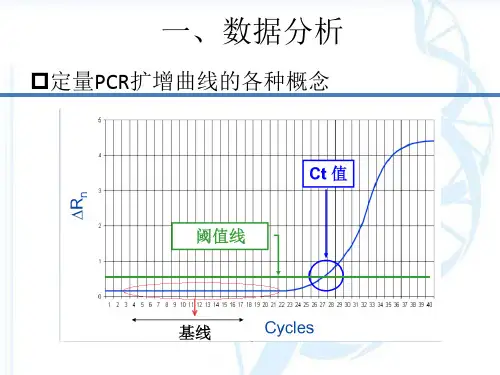
首先,荧光定量PCR数据的分析通常包括以下几个步骤:1.荧光PCR数据的获取:荧光定量PCR实验过程中,荧光信号会被记录下来。
对于每个样品,会获得一条荧光曲线,曲线上的荧光信号强度与PCR循环次数(Ct值)有关。
2. 荧光曲线的阈值设置:荧光曲线上的信号强度在实验的早期循环中较低,随着PCR循环的进行逐渐增加,直至达到一个稳定的平台。
阈值(threshold)是在这个平台上设置的一个信号强度的固定值,用于确定Ct值。
常用的阈值设置方法包括固定阈值法和导数阈值法。
3.Ct值的计算:Ct值是荧光定量PCR实验中的一个重要指标,表示荧光信号达到阈值的循环次数。
在荧光曲线上,Ct值可以通过与阈值的交点来确定。
Ct值越小,表示目标DNA序列的丰度越高。
4.样品之间的Ct值比较:荧光定量PCR实验中,通常需要同时检测一些内部参考基因(如GAPDH)作为对照,以便进行样品之间的Ct值比较。
内部参考基因应在不同样品中表达稳定,其Ct值应接近。
通过对目标基因的Ct值与内部参考基因的Ct值进行比较,可以计算出样品中目标DNA序列的相对丰度。
5.目标DNA序列的绝对丰度计算:通过构建标准曲线,可以将目标DNA序列的相对丰度转化为绝对丰度。
标准曲线是通过在实验中使用一系列已知浓度的目标DNA序列标准品进行绘制的。
通过测量标准品的Ct值并绘制荧光信号与目标DNA序列浓度的关系曲线,可以通过比较样品的Ct值与标准曲线来计算目标DNA序列的绝对丰度。

荧光定量PCR常见问题分析荧光定量PCR常见问题分析Q1.CT值出现过晚1.扩增效率低,反应条件不够优化。
设计更好的引物或探针;改用三步法进行反应;适当降低退火温度;增加镁离子浓度等。
2.PCR各种反应成分的降解或加样量的不足。
3.PCR产物太长。
一般采用80-150bp的产物长度。
Q2.无CT值(信号)出现1.反应循环数不够。
一般都要在35个循环以上,可根据实验情况增加循环(如至45cycles),但高于45个循环会增加过多的背景信号。
2.检测荧光信号的步骤有误。
一般SG法采用72℃延伸时采集,Taqman法则一般在退火结束时或延伸结束采集信号。
3.引物或探针降解。
可通过PAGE电泳检测其完整性。
4.引物或探针的设计,如探针高于引物的温度不够,造成探针未杂交上而产物已延伸的情况。
5.模板量不足。
对未知浓度的样品应从系列稀释样本的最高浓度做起。
6.模板降解。
避免样品制备中杂质的引入及反复冻融的情况。
Q3.标准曲线的线性关系不佳1.加样存在误差,使得标准品不呈梯度。
2.标准品出现降解。
应避免标准品反复冻融,或重新制备并稀释标准品。
3.引物或探针不佳。
重新设计更好的引物和探针4.模板中存在抑制物,或模板浓度过高Q4.阴性对照也出现明显的起飞1.应mix或水被污染。
2.引物二聚体的出现。
用SG法在35cycles以后阴性出现起飞属正常情况,可配合熔解曲线进行分析。
3.反应过程中探针的降解。
用PAGE电泳对探针进行检测。
4.如果使用了ROX校正,则可能是ROX的降解所造成。
Q5.在溶解曲线前是否插入一个同溶解程序初始温度相同的一个保温过程如果在熔解曲线前没有一个保温过程,则曲线会在前几个循环非常陡峭,使真正的熔解峰型几乎看不到。
Q6.熔解曲线不止一个主峰1.引物设计不够优化。
应避免引物二聚体和发夹结构的出现。
2.引物浓度不佳。
适当降低引物的浓度,并注意上下游引物的浓度配比。
3.镁离子浓度过高。
适当降低镁离子浓度,或选择更合适的mix试剂盒。

医学检验中实时荧光定量PCR技术的使用注意事项分析实时荧光定量PCR技术是一种广泛应用于医学检验领域的分子生物学技术,具有高灵敏度、高特异性和高度定量的特点。
在使用实时荧光定量PCR技术进行医学检验时,需要注意以下几点。
首先,实验室操作规范是保证实时荧光定量PCR技术准确性和可靠性的关键。
操作人员应该严格按照实验室的操作规程进行操作,并遵循好实验室规范。
操作前应进行充分的准备工作,包括准备实验所需试剂、标准品、阳性对照和负对照等。
同时,应根据实际需要设计好实验方案,包括引物和探针的选择以及扩增条件的优化等。
其次,实时荧光定量PCR技术的反应体系也需要特别注意。
在制备反应液时,应按照标准操作流程严格配制,确保各组分的准确浓度,避免误差的出现。
此外,在反应体系中还应添加可见染料以判断反应是否正常进行,在出现问题时及时调整和排除故障。
同时,反应体系的体积也需要根据不同的实验目的进行合理调整,以保证准确的定量结果。
第三,控制实时荧光定量PCR技术过程中的污染也是非常重要的。
PCR反应非常敏感,即使少量的外源DNA也可能对结果产生干扰。
因此,在样品的提取、纯化和扩增过程中,应当严格控制外源DNA的污染。
实验过程中应设有负对照组和阳性对照组,用于确认试剂和系统是否受到污染,以确保结果的可靠性。
此外,实验人员还应注意不要将样品或试剂带到其他实验室或工作区域,以免交叉感染。
此外,实时荧光定量PCR技术的数据分析也需要谨慎。
在实验过程中,所得到的荧光信号曲线应具有明确的指数增长阶段,并且有足够高的信噪比。
对于每个PCR反应,都需要做好内部质控,如负对照组和阳性对照组。
在数据分析时,应使用专业的软件进行数据处理和结果解读,避免主观因素对结果的干扰和误判。
同时,还需要将结果与临床实际情况进行综合判断,避免误诊或漏诊的发生。
最后,实时荧光定量PCR技术在医学检验中的应用也需要结合实际情况进行合理选择。
虽然实时荧光定量PCR技术具有高度灵敏性和特异性,但并不适用于所有的检测需求。


荧光定量PCR常见问题及解答更新時間:2010-12-10 10:43:37一、无 Ct 值(信号)出现:1.反应循环数不够:一般都要在 35 个循环以上,可根据实验情况增加循环,但高于45个循环会增加过多的背景信号;2.检测荧光信号的步骤有误:一般采用 72℃延伸时采集荧光,Taqman 方法则一般在退火结束或延伸结束采集信号;3.引物或探针降解:可通过PAGE电泳检测其完整性;4.探针设计不佳:设计探针温度低于引物,造成探针未杂交上而产物已延伸;5.模板量少:对未知浓度的样品应从系列稀释样品的最高浓度做起;6.模板降解:避免样品制备中杂质的引入以及模板反复冻融的情况发生。
二、如何确认模板中是否含有 PCR 反应阻害物质:有时RNA或cDNA模板中存在对反转录和荧光PCR 反应的阻害物质。
为了确认这样的阻害物质的有无,我们可以使用高浓度的模板按 3-4 个梯度稀释,并使用其进行荧光定量 PCR 反应。
如果无阻害物质存在,得到的 Ct 值就会依模板浓度的变化而变化;而如果模板中有阻害物质的存在,实验过程中我们就会发现高浓度的模板有反应性能下降的现象。
三、Ct值出现过晚:1.反应条件不佳:未最佳反应条件,可重新设计引物或探针,适当降低退火温度,增加镁离子浓度等;2.PCR各种反应成分的降解或加样量不够;3.扩增产物片段过长:一般采用 100-200bp 的扩增长度。
四、标准曲线的线性关系不佳:1.加样存在误差,标准品稀释不呈梯度;2.标准品降解:标准品尽量避免反复冻融;3.引物或探针不佳:重新设计;4.模板中存在抑制物,或模板浓度过高。
五、阴性对照液出现明显的扩增:1.荧光 PCR mix 或水被污染;2.引物二聚体的出现:在 35 循环后阴性对照出现扩增属正常情况,可配合溶解曲线进行分析;3.反应过程中探针降解:用PAGE电泳对探针进行检测。
六、溶解曲线不止一个主峰:1.引物设计不佳:避免二聚体和发夹结构的出现;2.引物浓度不佳:适当调整引物浓度;3.退火温度低:提高退火温度;4.镁离子浓度过高:适当降低镁离子浓度;5.模板中有基因组的污染:RNA提取过程中避免基因组 DNA 的污染,或通过引物设计避免非特异扩增。

荧光定量PCR实验及数据分析一、概述荧光定量PCR(Quantitative Realtime PCR,简称qPCR)是一种结合了PCR技术的高灵敏度和荧光探针技术的实时定量特性的分子生物学分析方法。
该方法通过实时监测PCR反应过程中荧光信号的变化,对模板DNA或RNA的初始浓度进行定量分析。
荧光定量PCR技术在基因表达研究、病原体检测、基因突变分析以及药物疗效评估等领域具有广泛的应用价值。
在荧光定量PCR实验中,通常使用特异性引物和荧光探针来识别并扩增目标序列。
荧光探针的设计是关键步骤之一,它必须能够与目标序列特异性结合并在PCR过程中产生可检测的荧光信号。
实验过程中还需严格控制反应条件,包括温度、时间、引物和探针的浓度等,以确保实验的准确性和可重复性。
数据分析是荧光定量PCR实验不可或缺的一部分。
通过对实验数据的收集、整理和分析,可以获取目标序列的初始浓度信息,进而对实验结果进行解读和评估。
数据分析方法包括相对定量和绝对定量两种,前者通过比较不同样本间目标序列的相对表达量来评估差异,后者则通过标准曲线法或质粒拷贝数法等方法来确定目标序列的绝对浓度。
荧光定量PCR技术是一种高效、灵敏且特异的分子生物学分析方法,对于研究基因表达、病原体检测等领域具有重要意义。
通过不断优化实验操作和数据分析方法,可以进一步提高荧光定量PCR实验的准确性和可靠性,为科学研究和临床实践提供有力支持。
1. 荧光定量PCR技术的概述荧光定量PCR技术,是一种基于DNA聚合酶链式反应的分子生物学技术,它通过引入荧光标记,实时监测PCR过程中目标DNA片段的扩增情况,从而实现对特定基因拷贝数的精确量化。
该技术结合了PCR的高效扩增能力与荧光信号的灵敏检测,使得微量DNA分子的检测成为可能,并在遗传学、分子生物学、医学诊断等领域中发挥着重要作用。
荧光定量PCR技术主要依赖于特异性引物和探针的设计,使得PCR扩增过程具有高度的特异性。


分析与检测Tlogy科技34 食品安全导刊 2017年4月昆山吉和力仪器有限公司(以下简称“吉和力”)是一家提供专业服务和仪器设备的集成供应商,公司以用户实际需求为导向,提供最合适、性价比最高的仪器,致力于打造一站式服务平台。
为了让业内人士对qPCR 进行更加深入的了解,吉和力整理了一些相关常见问题,并对这些问题一一进行了解答。
qPCR 的英文全称是Real-time Quantitative PCR Detecting System,即实时荧光定量核酸扩增检测系统,也叫实时定量基因扩增荧光检测系统,简称qPCR。
简单来说,qPCR 就是利用荧光信号的变化,实时监测PCR 扩增反应中每一个循环扩增产物量的变化,通过Ct 值和标准曲线的分析对起始模板进行定量分析。
而常规的PCR 无法对起始模板准确定量,无法对扩增反应实时监测。
如何提高RT-PCR 反应的灵敏度与特异性?①确定模板RNA 完整性,无DNA 污染;②RNA 模板中不应含有扩增反应抑制剂;③为了防止模板降解,在反应体系中加入RNase 抑制剂RNasin;④使用适量的模板RNA,模板量太多会降低特异性,太少会导致扩增不出条带或条带太弱;⑤若模板中有二级结构,可通过提高逆转录反应温度来提高扩增效果;⑥设计引物时,避免在引物3’端含有互补序列,避免形成内部发卡结构。
避免RNA 降解的方法有哪些?①在用来验证完整性之前先在变性胶上分析RNA;②运用良好无污染技术分离RNA;③将组织从动物体取出后立刻提取RNA,并将提取好的RNA 反转录为cDNA 进行低温保存。
RNA中含有逆转录抑制剂时,怎么处理?逆转录抑制剂包括:SDS、EDTA、甘油、焦磷酸钠、亚精胺和胍盐,可将对照RNA 与样品混合,同对照RNA 反应比较产量以检测RNA 抑制剂;若对照RNA 与样品混合后产量降低,则说明样品中存在逆转录抑制剂,可用70%(v/v)乙醇对RNA 沉淀进行清洗,以除去抑制剂。

荧光定量PCR常见问题解析荧光定量PCR常见问题解析(一)1、普通PCR与荧光定量PCR技术区别?①荧光定量PCR能够实时监测PCR反应的全过程,对每一个循环进行定量或定性分析;而普通PCR只能对反应的终产物进行半定量或定性分析。
②荧光定量PCR的结果分析可以直接通过电脑读出,而普通PCR 的结果必须通过下一步的电泳进行条带分析。
2、荧光定量PCR技术的特点①灵敏度高、特异性强。
② PCR实验过程全封闭,仪器自动分析和获取实验结果,无后续实验操作。
③实时监测反应过程,定量更准确。
④定量范围宽,可达到10个数量级。
⑤可以实现一次反应检测多个样本。
3、实时荧光定量PCR实验操作注意事项①应该带一次性乳胶手套并经常更换,在试剂准备室和样本处理室应该配备负压式生物安全柜,以防止对环境或对样品的污染。
②要严格防止气溶胶交叉污染。
采取的措施有:使用一次性带滤芯移液枪吸头;不使用吸头吹打液体进行混匀;在生物安全柜中进行离心操作;尽量减少在生物安全柜以外开启试剂或PCR管;禁止在荧光定量PCR结束后打开PCR管盖。
③实验中牵涉的有毒有害及有污染的样本及试剂应该严格按照生物安全相关规定操作和处理。
④荧光探针应该避光保存,加入反应液后应尽快上机,以防探针粹灭。
⑤在进行RNA相关操作时,必须使用无RNase的实验器具及耗材,并加样后立即上机操作,以防RNA的降解。
⑥所有试剂在开启之前都要瞬时离心;试剂配制完成后要瞬时离心以去除气泡。
⑦不要在荧光定量PCR管上做任何标记,不能用手接触PCR管盖上采光部位。
4、实时荧光定量PCR实验无CT值出现或无扩增信号的问题及解决方案①实验的循环数不够,设计的循环数应该在35以上,可增加至45个循环,但不可高于45个循环。
②通道选择不对,应该仔细阅读产品说明书,根据说明书的要求选择采光通道。
③荧光采集位置设计有误,应该仔细阅读产品说明书,根据说明书的要求设置荧光信号采集位置。
④引物或探针降解,要求厂家重新发货验证。
临床荧光PCR结果分析及常见问题临床荧光PCR结果分析及常见问题中国⼈民解放军三零⼆医院王海滨⼀、荧光 PCR 技术在临床诊、疗中的应⽤PCR 技术是于 1983 年由 Mullis 发明,后来传到我国,但是由于实验污染的问题, 1998 年卫⽣部取缔 PCR 实验⽅法在临床诊断中的应⽤。
荧光 PCR 出现后,污染减少很多,但近⼏年由于磁珠的应⽤,污染⼜再起。
2003 年, PCR 在 SARS 的早期诊断中⾮常有效,当时使⽤的是电泳法。
H1N1 甲型流感( 2009 ):荧光 PCR 技术作为确诊指标( 755 份 / 天)。
疾病易感基因的研究( SNP ): CR1/ 丙型肝炎 IL-28B 。
疾病(肿瘤)易感基因的鉴定。
⼆、影响临床荧光 PCR 检测结果的因素(⼀)影响临床荧光 PCR 检测结果的常见因素1. 检验因素:实验室硬件、仪器、试剂、⼈员素质、技术⽔平、管理⽔平。
2. ⾮检验因素:标本采集、质量、部位、标本保存、条件、时间、标本中的⼲扰物质、样本丢失!(⼆)加强检测前标本的质量管理——避免问题重复发⽣规范标本前处理流程、制度化的重要性,建⽴不合格标本记录和定期总结制度、制定解决措施与可⾏路径(编号和项⽬错误等),查找标本丢失可能的环节。
加强本室新进⼯作⼈员流程培训,经常性进⾏临床医护的交流与培训。
(三)实验室分区分为:试剂配制、核酸提取、 PCR 扩增以及开放产物分析等区域。
分区⽬的:通过物理分割达到标本、试剂或使⽤器具等被沾染或掺⼊污染,标本间的交叉污染等。
分区要求:⾄少要有清洁的试剂配制和核酸制备区三、试剂配制(⼀)试剂配制室的设置与维护以空间内⽆核酸⽓溶胶为准!1 .正压、清洁进风 ( 壁挂式空调 ) 。
试剂冰箱内勿存放病原或核酸相关物品!定期清洁除霜!2 .有效氯消毒液清洁实验室台⾯、地⾯。
3 .移液器:使⽤前的清洁。
4 .离⼼机等:每周定期⽊浆纸⼱擦拭,⽅法。
“⼀次性物品的使⽤ - 从 COBAS 的“浪费”中找到均衡!(⼆)试剂配制注意事项试剂配制实际是简单的问题,但是也是⾮常复杂的环节。
荧光实时定量PCR 结果分析问题荧光实时定量PCR结果分析问题2020-01-22 13:08Line Gene实时荧光定量PCR不同结果分析模式对检测结果的阻碍目的探讨实时荧光定量PCR不同结果分析模式对检测结果的阻碍。
方式用Line Gene FQD 33A型荧光定量PCR检测系统检测本院门诊和病房乙肝病人血清HBV DNA 77份,别离用最大二阶导数法和样点拟合法进行结果分析。
结果发觉两种分析方式的整体相关性较好(r=。
但HBV DNA拷贝数含量<105时,两种模式的相关性差(r=,现在,最大二阶导数法得出的数值要比样点拟合法低(t=,P<,且易显现假阴性结果。
结论尽管样点拟合法步骤较多,但其结果更靠得住,因此进行结果分析时,建议利用样点拟合法。
而且当样本中HBV DNA拷贝数<105时只能利用样点拟合法进行结果分析。
实时荧光定量;聚合酶链反映;质量操纵7.数据分析:(1)相对定量:利用△Ct法计算目的基因在不同组间的表达不同。
(2)绝对定量:将样品的Ct值代入标准曲线,以对目的基因进行定量。
(3)熔解曲线分析:确信产物特异性。
要紧实验步骤:1.设计并合成Real time PCR引物。
2.引物溶解后利用Promega的TaqDNA聚合酶进行含SG(SYBRGreen)的PCR反映优化。
3.客户样品RNA抽提。
a.实验对象为组织样品,取适量(50-100mg)新鲜组织样品或正确保留的组织样品,加1ml的RNA抽提试剂Trizol(Invitrogen),匀浆后抽提RNA。
b.实验对象为细胞样品,每份样品取1×106~1×107细胞,PBS清洗细胞,去PBS加1ml的RNA抽提试剂Trizol(Invitrogen),裂解后抽提RNA。
质量检测a.紫外吸收测定法测定RNA在分光光度计260nm和280nm处的吸收值,以计算其浓度并评估RNA纯度。
b.利用ambion公司的甲醛电泳试剂进行变性琼脂糖凝胶电泳,检测RNA纯度及完整性。