荧光定量PCR_完整版
- 格式:pdf
- 大小:1.55 MB
- 文档页数:22



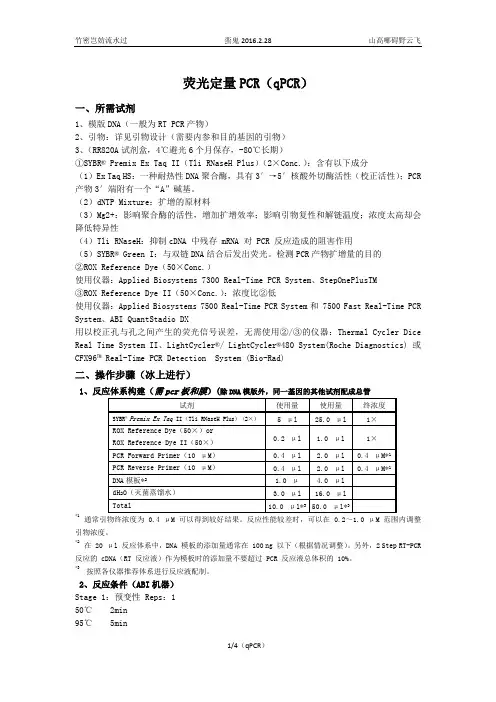
荧光定量PCR(qPCR)一、所需试剂1、模版DNA(一般为RTPCR产物)2、引物:详见引物设计(需要内参和目的基因的引物)3、(RR820A试剂盒,4℃避光6个月保存,-80℃长期)①SYBR® Premix Ex Taq II(TliRNaseH Plus)(2×Conc.):含有以下成分(1)Ex Taq HS:一种耐热性DNA聚合酶,具有3′→5′核酸外切酶活性(校正活性);PCR 产物3′端附有一个“A”碱基。
(2)dNTP Mixture:扩增的原材料(3)Mg2+:影响聚合酶的活性,增加扩增效率;影响引物复性和解链温度;浓度太高却会降低特异性(4)TliRNaseH:抑制cDNA 中残存 mRNA 对 PCR 反应造成的阻害作用(5)SYBR® Green I:与双链DNA结合后发出荧光。
检测PCR产物扩增量的目的②ROX Reference Dye(50×Conc.)使用仪器:Applied Biosystems 7300 Real-Time PCR System、StepOnePlusTM③ROX Reference Dye II(50×Conc.):浓度比②低使用仪器:Applied Biosystems7500 Real-Time PCR System和 7500 Fast Real-Time PCR System、ABI QuantStadio DX用以校正孔与孔之间产生的荧光信号误差,无需使用②/③的仪器:Thermal Cycler Dice Real Time System II、LightCycler®/ LightCycler®480 System(Roche Diagnostics) 或CFX96™ Real-Time PCR Detection System (Bio-Rad)二、操作步骤(冰上进行)1、反应体系构建()(除DNA模版外,同一基因的其他试剂配成总管*1通常引物终浓度为 0.4 μM 可以得到较好结果。



附录1第一步:RNA的提取材料:灭菌过的枪头、匀浆器、EP管、去离子水;TRIzol Reagent、三氯甲烷、异丙醇、、无水乙醇、离心机。
步骤:1)将大约100mg组织加入研磨器,加入1ml的TRLzol Rengent研磨20-30min;直到看不到大块的组织。
2)将液体倒入灭菌后的EP管内,加三氯甲烷250 μl,上下颠倒充分混匀,静置5min,4℃离心,13000×g,10min。
3)取上清液500 μl于另一灭菌过的EP管内,加入等量的异丙醇,混匀后放入4℃的冰箱15min后取出,4℃离心,13000×g,10min;倾倒出上面的液体,留在管底的白色沉淀即为RNA。
5)加入75%的乙醇1ml,4℃离心,7500×g,5min。
6)将上面的液体倒出,只剩白色沉淀;再瞬时离心,尽量吸干管内的液体,晾干,再加入适量的去离子水加以溶解。
第二步逆转录准备材料:逆转录试剂盒(TOYOBO,日本)反应体系:5×RT Buffer 4 μldNTP 2 μlOliga(dT)20 1 μlRNase free H2O 1μlRnase inhibitor 1μlReverTra Ace 1μlRNA 10μl总体系20μl 反应条件:42℃20min95℃5min4℃5min第三步Real-time PCR材料:SYBR Green mix(TOYOBO公司,日本)、上下游引物、cDNA、ddH2O 步骤:1、反应体系SYBR Green mix 12.5μlPlus solution 2.5μl上游引物(10pmol/μl)1μl下游引物(10pmol/μl)1μlddH2O 5.5μlcDNA(10倍稀释) 2.5μl总体系25μl2、反应条件50℃2min;95℃2min95℃15s Array退火15s72℃45s72℃10min;3. 扩增程序图:。


荧光定量PCR(Quantitative Real-Time PCR,简称qPCR)是一种分子生物学技术,用于精确测定样本中特定核酸序列的数量。
其基本原理基于PCR(聚合酶链式反应)技术和实时荧光检测,能够在PCR扩增过程中连续监测荧光信号的变化,从而实现对起始模板量的定量分析。
荧光定量PCR原理简述:1.PCR扩增:qPCR采用传统的PCR方法,包括变性(DNA双链解开成单链)、退火(引物与靶序列配对)和延伸(DNA聚合酶合成新链)这三个基本步骤,反复进行使得目标序列指数级扩增。
2.荧光标记与检测:SYBR Green法:SYBR Green是一种非特异性的双链DNA结合染料,在游离状态下几乎不发出荧光,但一旦与双链DNA结合后,荧光强度显著增强。
因此,随着PCR过程中的产物增加,荧光信号也相应增加,荧光强度与PCR产物的数量成正比。
TaqMan探针法:此方法更为特异,使用一种特殊的寡核苷酸探针,其两端分别标记了荧光报告基团和淬灭基团。
在PCR反应中,当探针与靶序列配对时,位于中间的探针被Taq 酶水解,导致荧光报告基团与淬灭基团分离,从而产生荧光信号。
只有当特定的扩增产物生成时才会释放荧光。
荧光定量PCR实验步骤概览:1.样品制备:RNA提取:从组织、细胞或其他生物样本中提取总RNA,常用TRIZOL或类似试剂进行裂解、离心分相和乙醇沉淀来纯化RNA。
cDNA合成:对于mRNA的定量,需要先将RNA逆转录为cDNA。
2.设计与合成引物:针对目标基因设计一对特异性的PCR引物,用于扩增目的片段。
3.PCR反应体系构建:将纯化的cDNA或DNA模板、特异性引物、Taq聚合酶、缓冲液、dNTPs和其他必要成分如SYBR Green染料或TaqMan探针等加入至PCR管中,配置成最终的PCR反应体系。
4.实时荧光PCR扩增与检测:在荧光定量PCR仪上进行PCR反应,仪器在每次循环的适当阶段收集荧光信号,并记录下来。

万字长文讲清荧光定量pcr荧光定量PCR(Fluorescent Quantitative Polymerase Chain Reaction,qPCR)是一种基于PCR技术的快速、高灵敏度和高特异性的DNA 定量方法。
它利用DNA扩增和荧光探针结合的原理,可以快速准确地定量目标DNA的含量。
荧光定量PCR主要分为两个步骤:扩增和检测。
在扩增步骤中,通过PCR反应使目标DNA被放大成大量的拷贝,并引入荧光标记的引物。
在检测步骤中,通过荧光探针与扩增产物结合,并测量荧光信号的强度,从而确定目标DNA的含量。
具体步骤如下:1. 反应体系的准备:根据实验需要,制备PCR反应液,包括引物、荧光探针、缓冲液、dNTPs、酶等。
2. DNA模板的准备:从待测样本中提取DNA,并进行纯化和浓缩。
3. PCR扩增反应:将DNA模板与引物和荧光探针加入PCR反应液中,进行PCR扩增反应。
PCR反应可以选择不同的温度程序,包括变性、退火和延伸等步骤。
4. 荧光信号检测:在PCR扩增反应过程中,荧光探针与目标DNA 结合形成双链结构,荧光信号被激发并发射。
荧光信号可以通过荧光实时定量PCR仪器进行检测和记录。
5. 数据分析:根据荧光信号的强度,可以计算目标DNA的含量。
通常,荧光信号的强度与目标DNA的初始浓度成正比。
荧光定量PCR的优势在于其高灵敏度、高特异性和高准确性。
由于荧光信号可以实时检测和记录,所以可以在PCR反应过程中实时监测目标DNA的含量变化。
此外,荧光定量PCR可以同时检测多个目标DNA,并且可以定量非常低浓度的DNA样本。
荧光定量PCR在生物医学研究、临床诊断、基因表达分析等领域得到了广泛的应用。
它可以用于检测病原体、基因表达水平、突变等,对于研究疾病的发生机制、筛选药物靶点、预测疾病进展等具有重要意义。
同时,荧光定量PCR也是一种常用的基因分型方法,可以用于DNA指纹鉴定、亲子鉴定等。
荧光定量PCR作为一种快速、高灵敏度和高特异性的DNA定量方法,已经成为生命科学研究和临床诊断的重要工具。

一、样品RNA的抽提1.匀浆将-80℃冻结的RNA提取样品迅速转移至用液氮预冷的研钵中,用研杵研磨组织,其间不断加入液氮,直至研磨成粉末状,向研钵中加入与样品匀浆量匹配的适量的RNAiso Plus,并将组织样粉末覆盖。
2.抽提室温放置5min,使核蛋白复合物完全溶解后转移至1.5mL无酶离心管中,室温静置5min,12000r/min,4℃离心15min;小心吸取上清液移入新离心管,加上清液的1/5体积量的氯仿,静置10min;12000 r/min,4℃离心10min。
(此时分三层:无色上清液、白色蛋白层、带颜色的下层有机相);3.RNA沉淀吸取上清液移至新离心管,切勿吸入中间蛋白层,向上清液中加入与上清液等量体积的异丙醇。
颠倒混匀后室温静置10min,12000r/min,4℃离心5min (此时底部会出现沉淀);4.RNA洗涤移去上清液,每1mlTRIZOL试剂裂解的样品中加入1 ml的75%乙醇(用DEPC水或无酶无菌水配制,超净台中配制),清洗RNA沉淀。
混匀后,4℃下12000r/min离心5分钟。
5.RNA溶解吸去大部分乙醇溶液,使RNA沉淀在室温空气中干燥5-10分钟。
加入无RNA酶的水20-40μL用枪反复吹打几次,使其完全溶解,获得的RNA溶液保存于-80℃待用。
二、RNA浓度和质量的测定1. 1%-1.2%琼脂糖电泳检测RNA的完整性:将从肌肉中提取的总RNA吸取5μL,加入1μL 6 X Loading Buffer,混匀,点入1%琼脂糖凝胶点样孔中,120V电压电泳,经凝胶成像系统检测条带。
2. 核酸蛋白分析仪检测OD;吸取2μL的RNA检测OD260/OD280检测含量与纯度,A260/A280在1.9-2.1之间,说明提取的总RNA质量很好,记录稀释后RNA的Conc值(浓度)。
三、实时定量引物实时定量引物根据NCBI公布的基因序列,依据实时定量引物设计原则,使用软件Primer5.0设计,管家基因引物引用参考文献所列,引物由上海生工有限公司合成。
荧光定量pcr1、总RNA抽提(*头和离心管均经过湿热灭菌,无RNA酶)1)取匀浆器,加入1ml的Trizol Reagent,置冰上预冷。
2)取100mg组织,加入到匀浆器中。
3)充分研磨直至无可见组织块。
4)13000g离心10min取上清。
5)加入250 μl三氯甲烷,颠倒离心管15s,充分混匀,静置3min。
6)4℃下13000g离心8min。
7)将上清转移到一新的离心管中,加入0.8倍体积的异丙醇,颠倒混匀。
8)-20℃放置15min。
9)4℃下13000g离心10min,管底的白色沉淀即为RNA。
10)吸除液体,加入75%乙醇1.5ml洗涤沉淀。
11)4℃下13000g离心5min。
12)将液体吸除干净,将离心管置于超净台上吹3min。
13)加入20μl无RNA酶的水溶解RNA。
14)55℃孵育5min。
2、反转录(*头和PCR均经过湿热灭菌,无RNA酶)1)取一PCR管,加入含2μg RNA的溶液。
2)加入1μl oligo(dT)15。
3)用无核糖核酸酶的去离子水补足至12μl。
4)于PCR仪上70℃保温5min,迅速置冰上冷却。
5)依次加入4μl 5×buffer,2μl 10mM dNTPs,1μl RNA inhibitor和1μl 反转录酶,用*抽吸混匀。
6)于PCR仪上42℃保温60min,结束后80℃保温5min灭活反转录酶。
3、定量PCR1)取0.2ml PCR管,配制如下反应体系,每个反转录产物配制3管。
2× qPCR Mix 12.5μl7.5μM基因引物2.0μl反转录产物 2.5μlddH2O 8.0μl2)取0.2ml PCR管,配制如下反应体系,每个反转录产物配制3管。
2×qPCR Mix 12.5μl7.5μM内参引物2.0μl反转录产物 2.5μlddH2O 8.0μl3)PCR扩增预变性 95℃,10min循环(40次) 95℃,15s→60℃,60s溶解曲线 75℃→95℃,每20s升温1℃4、结果处理ΔΔCT法:A=CT(目的基因,待测样本)- CT(内标基因,待测样本) B=CT(目的基因,对照样本)- CT(内标基因,对照样本) K=A-B表达倍数=2-K注意事项1、长度:15—30bp。