固定污染源排气中丙烯腈的测定-气相色谱法 HJT 37-1999
- 格式:doc
- 大小:118.00 KB
- 文档页数:8



气相色谱法测定固体废物中丙烯腈的方法研究气相色谱法是一种常用于分析和测定有机化合物的方法。
在测定固体废物中丙烯腈的方法研究中,通过气相色谱仪的使用,可以进行样品的布置、提取和分析,具有操作简单、准确度高的优点。
本文将介绍气相色谱法测定固体废物中丙烯腈的方法研究。
样品的准备是整个分析过程的关键。
固体废物中丙烯腈的提取需要先将样品进行粉碎和混合,以保证样品的均匀性。
然后,将样品加入合适的溶剂中进行提取,通常可以选择乙酸乙酯或四氯化碳等有机溶剂。
加入足够量的溶剂以使样品完全溶解,并充分摇动或超声处理,加速提取过程。
接下来,对提取液进行过滤以去除固体颗粒和杂质。
采用滤纸或其他过滤设备进行过滤,确保提取液的干净和纯度。
然后,将提取液进行进一步的浓缩或纯化。
可以使用旋转蒸发器或其他适当的方法将溶剂去除或浓缩,得到纯净的丙烯腈样品。
使用气相色谱仪对样品进行分析和测定。
将样品溶解在适当的溶剂中,注入气相色谱仪的进样口。
选择合适的色谱柱和色谱条件,设置使用的检测器。
常用的检测器包括火焰离子化检测器(FID)和电子捕获检测器(ECD)。
通过控制进样量、柱温和流动速率等参数,进行样品分离和定量分析。
参照标准曲线或内标法,计算出样品中丙烯腈的含量。
需要注意的是,在进行气相色谱分析时,要严格控制仪器的操作条件,以保证结果的准确性和重现性。
对于固体废物样品的测定,还需要注意样品的前处理和提取过程,以保证提取效果和样品的代表性。
气相色谱法是测定固体废物中丙烯腈含量的可靠方法。
通过样品的准备、提取、浓缩和分析过程,可以得到准确的结果。
在实际应用中,还可以结合其他分析方法进行验证和比较,以提高测定结果的可靠性。

气相色谱法测定固体废物中丙烯腈的方法研究气相色谱法是一种常用的分离和定量分析技术,可以用于测定固体废物中丙烯腈的含量。
本文将介绍气相色谱法测定固体废物中丙烯腈的方法研究。
为了提取固体废物中的丙烯腈,可以使用溶剂萃取法。
将固体废物样品与适量的有机溶剂(如二氯甲烷)混合,进行振荡或搅拌,使丙烯腈溶解到有机溶剂中。
然后,将溶剂中的丙烯腈分离出来,可以采用离心沉淀法或过滤法。
得到的溶液即可用于气相色谱的分析。
接下来,需要选择合适的色谱柱和气相色谱仪。
对于丙烯腈的分析,一般常用非极性色谱柱,例如HP-5、DB-5或DB-1等。
而对于气相色谱仪,则需要具备温度控制和检测器等功能。
在样品进样前,可以对溶液进行一定的预处理。
可以使用氮气吹干或浓缩溶剂,以确保样品不含有剧烈挥发的物质。
还可以采用浓缩溶液的方法,使得样品中的丙烯腈浓度达到分析的要求。
样品的进样方式可以选择自动进样器或手动进样。
若样品量较少,可以采用手动进样的方式,而样品量较多时,建议使用自动进样器,以提高分析效率。
在进行气相色谱分析时,需要选择适当的色谱条件。
对于非极性色谱柱,可以选择温度梯度或等温的程序升温。
温度的选择要考虑到丙烯腈的挥发性和色谱柱的热稳定性。
检测器可以选用多种类型,如火焰离子化检测器(FID)、氮磷检测器(NPD)等。
不同检测器的选择将影响到定量的灵敏度和选择性。
为了进行定量分析,需要建立标准曲线。
可以使用已知浓度的丙烯腈标样,通过检测器测量其峰面积,并与其浓度进行关联。
然后,通过测量待测样品的峰面积,利用标准曲线进行定量计算,得到丙烯腈的含量。
气相色谱法是一种可靠的方法,适用于测定固体废物中丙烯腈的含量。
通过适当的预处理、进样方式、色谱条件以及建立标准曲线,可以准确地测定固体废物中丙烯腈的含量,为固体废物管理和环境保护提供参考。



1适用范围1.1本标准适用于固定污染源有组织排放和无组织排放的丙烯腈测定。
1.2当采样体积为30L时,方法的检出限为0.2 mg/m3。
方法的定量测定浓度范围为0.26~33.0 mg/m3。
2方法原理丙烯腈(CH二CHCHCN)用活性炭常温吸附富集,再经二硫化碳常温解吸,解吸液中各组分通过色谱柱得到分离后进人氢火焰离子化检测器(FID),从测得的丙烯腈色谱峰高(或面积),对解吸液中丙烯腈浓度定量,最后由解吸液体积、浓度和采样体积计算出气体样品中丙烯腈的浓度。
3引用标准下列标准所包含的条文,通过在本标准中引用而构成为本标准的条文:GB 16297-1996 大气污染物综合排放标准GB 16157-1996 固定污染源排气中颗粒物测定和气态污染物采样方法4试剂和材料4.1丙烯腈:色谱纯(或分析纯,但必须对丙烯腈无色谱干扰峰)。
4.2二硫化碳:分析纯(对丙烯腈的色谱测定无干扰峰,否则需进行蒸馏,取46~47℃的馏分)。
4.3气相色谱固定相:GDX-502, 60~80目。
4.4氮气:纯度99.99%,并用分子筛或活性氧化铝净化.4.5氢气:纯度99.99%,并用分子筛或活性氧化铝净化。
4.6空气。
4.7活性炭吸附管活性炭吸附管的结构如图1所示。
玻璃管的两端熔封密闭,并配有两个塑胶帽盖,以备采样完毕后盖紧密闭用。
管内填装活性炭粒度为20〜40目4段含100mg, B段含50mg。
A段活性炭前的玻璃棉上压着一个V字型弹簧钩,以免炭粒松动。
活性炭应对气态丙烯腈有很强的吸附能力,并可用二硫化碳解吸被吸附的丙烯腈。
目前市售的用于采集空气中有机蒸气,并以二硫化碳作解吸溶剂的活性炭吸附管能满足要求。
4.8丙烯腊标准储备液:c=10. 0 mg/ml。
用分析天平准确称取一定t的丙烯腈(4.1)于容量瓶中,小心加人二硫化碳至刻度,配制成溶液的丙烯腈浓度为10.0 mg/ml,作为储备液,密闭存放于低温(4~8℃)下,备用。

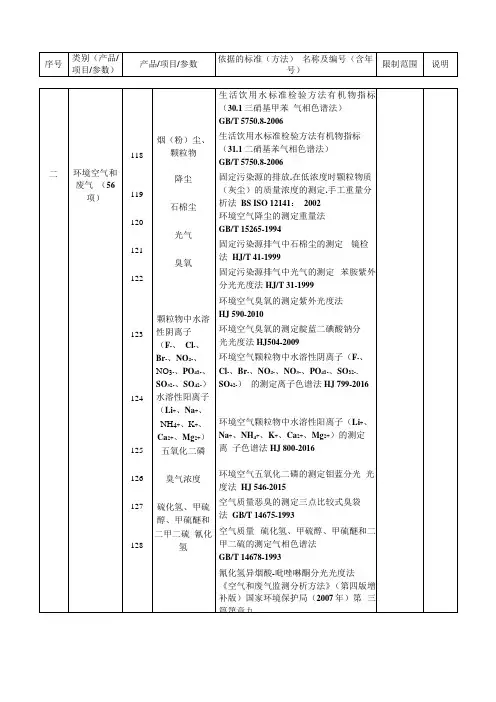
环境空气中VOCs检测的标准及环境空气检测分类环境空气中VOCs检测在环境检测的基本项目之一,地位自不用多说,所以,做好VOCs检测就成了必然一:定义VOCs是挥发性有机化合物(volatileorganic compounds)的英文缩写。
关于VOC的定义,不同的标准有不同的定义。
1.美国ASTM D3960-98标准将VOC定义为任何能参加大气光化学反应的有机化合物。
美国联邦环保署(EPA)的定义:挥发性有机化合物是除CO、CO2、H2CO3、金属碳化物、金属碳酸盐和碳酸铵外,任何参加大气光化学反应的碳化合物。
2.世界卫生组织(WHO,1989)对总挥发性有机化合物(TVOC)的定义为,熔点低于室温而沸点在50~260℃之间的挥发性有机化合物的总称。
3.有关色漆和清漆通用术语的国际标准ISO 4618/1-1998和德国DIN 55649-2000标准对VOC的定义是,原则上,在常温常压下,任何能自发挥发的有机液体和/或固体。
同时,德国DIN 55649-2000标准在测定VOC含量时,又做了一个限定,即在通常压力条件下,沸点或初馏点低于或等于250℃的任何有机化合物。
4.巴斯夫公司则认为,最方便和最常见的方法是根据沸点来界定哪些物质属于VOC,而最普遍的共识认为VOC是指那些沸点等于或低于250℃的化学物质。
所以沸点超过250℃的那些物质不归入VOC的范畴,往往被称为增塑剂。
这些定义有相同之处,但也各有侧重:如美国的定义,对沸点初馏点不作限定,强调参加大气光化学反应。
不参加大气光化学反应的就叫作豁免溶剂,如丙酮、四氯乙烷等。
而世界卫生组织和巴斯夫则对沸点或初馏点作限定,不管其是否参加大气光化学反应。
国际标准ISO 4618/1-1998和德国DIN 55649-2000标准对沸点初馏点不作限定,也不管是否参加大气光化学反应,只强调在常温常压下能自发挥发。
甲醛也是挥发性有机化合物,但甲醛易溶于水,与其他挥发性有机化合物有所不同,室内来源广泛,释放浓度也高。

丙烯腈①固定污染源排气中丙烯腈的测定气相色谱法(HJ/T37-1999)①丙烯腈的测定气相色谱法《空气和废气监测分析方法》(第四版)一、填空题1.气相色谱法测定环境空气或废气中丙烯腈时,使用检测器进行测定。
①①答案:氢火焰离子化(FID)2.《固定源污染源排气中丙烯腈的测定气相色谱法》(HJ/T 37-1999)的原理是:丙烯腈经吸附采样管常温富集后,用在常温下解吸,经色谱柱分离,气相色谱测定。
①①答案:活性炭二硫化碳3.气相色谱法测定环境空气或废气中丙烯腈,分析成分复杂样品且对定性结果有疑问时,可采用色谱柱定性。
若对定性结果仍有疑问时,可采用等其他方法和手段进一步定性。
①①答案:双气相色谱/质谱4.根据《固定源污染源排气中丙烯腈的测定气相色谱法》(HJ/T 37-1999)测定固定污染源排气中丙烯腈,在排气筒采集样品时,应在采样管口处塞适量的,然后将其伸入排气筒内的采样点位置,注意先使排气筒内的气体由吸附管流通,以充分洗涤采样管路,然后再使排气通过吸附管。
①答案:玻璃棉旁路5.气相色谱法测定环境空气或废气中丙烯腈时,采样后的活性炭管,在两端塞紧帽盖的情况下保存,低温(8℃以下)下最多可存放 d。
①①答案:避光 7二、判断题1.气相色谱法测定环境空气或废气中丙烯腈时,填充剂为80~100目GDX-502的色谱柱对丙烯腈和干扰物的分离效果要好于填充60~80目GDX-502的色谱柱。
( )答案:正确2.二硫化碳的沸点较低,易挥发,因此在配制丙烯腈标准溶液和对样品进行解吸时,注意随时盖紧容器的磨口塞,室内温度不要超过35℃。
( )①①答案:错误正确答案为:室内温度不要超过30℃。
3.气相色谱法测定环境空气或废气中丙烯腈时,采样装置连接好后,应检查系统的气密性和可靠性,并且整个采样装置的连接管要尽可能短。
( )①①答案:正确三、选择题1.采集固定源污染源排气中丙烯腈所用的活性炭粒度为目。
( )①A.20~40 B.40~60 C.60~80 D.80~100答案:A2.气相色谱法测定环境空气或废气中丙烯腈时,所采用的色谱柱填充剂为目的GDX-502,若遇到干扰物质量稍大而影响丙烯腈出峰时,可改为目的GDX-502。

气相色谱法测定固体废物中丙烯腈的方法研究随着现代工业的快速发展,固体废物已经成为了环境保护面临的重要问题之一。
其中,很多固体废物中含有丙烯腈,它可以对身体造成严重的危害。
因此,急需研究一种测定固体废物中丙烯腈含量的方法,从而更好地为环境保护提供有力的监管手段。
本文将介绍使用气相色谱法测定固体废物中丙烯腈的方法研究。
1、实验原理气相色谱法是一种分离和分析混合气体组分的方法。
该方法主要基于混合气体组分在分离栏中的相对亲和力的不同而实现气体组分的分离。
利用这种方法可以进行高效、灵敏和专门的定量分析。
对于固体废物中的丙烯腈含量的测定,可以通过气相色谱法进行检测。
具体来说,使用气相色谱仪将样品中的丙烯腈蒸发并吸附到实验柱上,然后利用柱中两相相互交互的条件,将各种气体组分分离,从而实现丙烯腈的分离和测定。
2、实验步骤2.1 样品预处理收集需要测定的固体废物,进行干燥处理,去除其中的水分和杂质。
然后将样品进行粉碎处理,使其得到均匀颗粒大小的样品。
2.2 抽提过程取出约1g的固体废物样品,加入一定量的丙酮(10ml),放入中温水浴中加热,直到样品中丙烯腈完全溶解。
将过滤后的液体置于闪蒸器中,加入少量的纯水,接着用干净的气相吸附管进行吸附,得到丙烯腈的吸附样品。
2.3 气相色谱分析将吸附到气相吸附管中的样品移植至气相色谱仪中进行分析。
在气相色谱仪中,使用柱色谱分离,然后可通过检测器测定并计算出丙烯腈的含量。
3、实验结果通过实验验证,上述方法可以非常有效地测定固体废物中丙烯腈的含量。
使用此方法,可以得到准确、可重复的数据,同时还可以提高分析效率,降低成本。
具体来说,通过测定的数据,可以用来指导有关部门制定环保方针,以及为现有固体废物环境卫生治理提供参考意见。
总之,气相色谱法测定固体废物中丙烯腈的方法研究,可以很好地帮助我们解决固体废物环境卫生方面的问题。
为生态环保事业贡献一份力量。
丙烯腈排放标准规范丙烯腈(Poly acetylenoleamine)是一种有机化合物,分子式为 CHO,分子量约为267.15。
丙烯腈在常温下不溶于水,不溶于乙醚、苯、氯仿等有机溶剂,但能溶解于浓硝酸和浓硫酸中。
丙烯腈气相色谱法(GC-MS)测定。
色谱柱为 HPLC;进样量25μ l;进样量25μ l,气相中含100 mg/L的丙烯腈。
一、适用范围丙烯腈主要用于制造尼龙66的原料,也用于生产聚酰胺纤维和聚酯,还可用于医药和农药中间体。
该物质主要危害人体健康,对水体、土壤、大气环境及其他环境均有污染,属于环境优先控制化学品。
1.[危险性类别]:第2.3类,第1类毒害品(第一类);2.[毒理学资料]:急性毒性:大鼠经口4千毫克/公斤;经皮肤接触0.2毫克/kg;3.[特殊毒性]:大鼠经口40千克,致突变性研究证明其致畸、致癌;4.[生产和使用控制]:生产过程应在密闭系统中进行。
5.[环境保护措施]:污水处理系统应配备足够的新鲜、干净、完整的饮用水。
6.[卫生防护措施]:车间人员工作时,必须穿戴工作服,戴口罩;接触高浓度作业时应佩戴防护眼镜和防毒口罩。
7.[安全技术措施]:工作场所应保持良好的通风,操作人员应注意个人卫生。
8.[消防措施]:雾状水灭火、泡沫灭火。
9.[个体防护措施]:穿戴化学防护用具(如防护镜及防护服),用水雾和干粉灭火剂扑救火灾。
10.[废弃处置方法]:焚烧或者掩埋。
二、规范性引用文件下列文件中的条款通过本规范的引用而成为本规范的条款。
凡是注日期的引用文件,其随后所有的修改单(不包括勘误的内容)或修订版均不适用于本规范,然而,鼓励根据本规范达成协议的各方研究是否可使用这些文件中的条款。
凡是不注日期的引用文件,其最新版本适用于本规范。
GB/T 18883-2002挥发性有机物排放控制标准,第4.1.14条规定:本标准所用全部引文均为已出版或修订过的标准。
使用该等引文时,应参照原发布的标准。
环境空气中VOCs检测的标准及环境空气检测分类环境空气中VOCs检测在环境检测的基本项目之一,地位自不用多说,所以,做好VOCs检测就成了必然一:定义VOCs是挥发性有机化合物(volatileorganic compounds)的英文缩写。
关于VOC的定义,不同的标准有不同的定义。
1.美国ASTM D3960-98标准将VOC定义为任何能参加大气光化学反应的有机化合物。
美国联邦环保署(EPA)的定义:挥发性有机化合物是除CO、CO2、H2CO3、金属碳化物、金属碳酸盐和碳酸铵外,任何参加大气光化学反应的碳化合物。
2.世界卫生组织(WHO,1989)对总挥发性有机化合物(TVOC)的定义为,熔点低于室温而沸点在50~260℃之间的挥发性有机化合物的总称。
3.有关色漆和清漆通用术语的国际标准ISO 4618/1-1998和德国DIN 55649-2000标准对VOC的定义是,原则上,在常温常压下,任何能自发挥发的有机液体和/或固体。
同时,德国DIN 55649-2000标准在测定VOC含量时,又做了一个限定,即在通常压力条件下,沸点或初馏点低于或等于250℃的任何有机化合物。
4.巴斯夫公司则认为,最方便和最常见的方法是根据沸点来界定哪些物质属于VOC,而最普遍的共识认为VOC是指那些沸点等于或低于250℃的化学物质。
所以沸点超过250℃的那些物质不归入VOC的范畴,往往被称为增塑剂。
这些定义有相同之处,但也各有侧重:如美国的定义,对沸点初馏点不作限定,强调参加大气光化学反应。
不参加大气光化学反应的就叫作豁免溶剂,如丙酮、四氯乙烷等。
而世界卫生组织和巴斯夫则对沸点或初馏点作限定,不管其是否参加大气光化学反应。
国际标准ISO 4618/1-1998和德国DIN 55649-2000标准对沸点初馏点不作限定,也不管是否参加大气光化学反应,只强调在常温常压下能自发挥发。
甲醛也是挥发性有机化合物,但甲醛易溶于水,与其他挥发性有机化合物有所不同,室内来源广泛,释放浓度也高。
气相色谱法测定固体废物中丙烯腈的方法研究【摘要】气相色谱法是一种常用的分析方法,本研究旨在探讨该方法在固体废物中测定丙烯腈的应用。
通过对样品前处理方法的优化和仪器条件的合理设置,实现了对丙烯腈的快速准确测定。
实验结果表明,该方法具有较高的灵敏度和重现性,能够有效地应用于固体废物中丙烯腈的检测。
在实验结果分析中发现,该方法具有一定的检测误差,但总体上仍具有较高的可靠性。
未来,可以进一步完善该方法,提高检测精度和准确性,以满足不同领域对丙烯腈检测的需求。
本研究对于推动固体废物中丙烯腈的监测和治理具有一定的参考意义。
【关键词】气相色谱法、固体废物、丙烯腈、样品前处理、仪器条件、实验操作、数据处理、实验结果、优缺点、未来展望1. 引言1.1 研究背景本研究旨在探讨利用气相色谱法测定固体废物中丙烯腈的方法,旨在建立一种快速、准确的检测方法,为固体废物中有害成分的监测提供技术支持,并为环境保护和人体健康提供科学依据。
这将有助于加强对固体废物处理的监管与管理,减轻环境污染带来的危害,提高废物资源化利用效率。
1.2 研究目的本研究的目的是通过气相色谱法对固体废物中丙烯腈的测定方法进行研究,探讨在固体废物处理过程中可能存在的丙烯腈污染问题。
通过建立有效的分析方法,可以准确快速地检测固体废物中丙烯腈的含量,为环境监测和废物处理提供科学依据。
通过本研究也可以为气相色谱法在固体废物分析中的应用提供一定参考。
通过本研究,旨在建立一套准确、快速、可靠的气相色谱法分析方法,为固体废物中丙烯腈的检测提供技术支持和方法参考。
通过实验结果的分析和总结,进一步探讨该方法的优缺点,为今后的相关研究提供参考和借鉴,推动固体废物中有害物质的监测和治理工作。
1.3 研究意义本研究的意义主要体现在以下几个方面:固体废物中丙烯腈的测定方法的研究,有助于监测和控制工业生产过程中可能产生的有害化学物质排放。
丙烯腈是一种剧毒化合物,对人体健康和环境造成严重危害。
水中丙烯腈的测定气相色谱法
气相色谱法用于测定水中的丙烯腈。
气相色谱是一种无毒、灵敏、快速的分析测试方法,常用于分析气态样品,也可以应用于液态和固态样品的分析。
空气中含有丙烯腈,以及水中含有丙烯腈,但它们的分析和测试过程不同。
测定水中丙烯腈时,必须使用气相色谱法。
用气相色谱法测定水中丙烯腈的步骤如下:
1、准备和操作:准备一台配备气相色谱分析仪的实验台、一台潜水泵、一组测定装置,其中包括池子、一台压力传感器、一台校准器。
2、方法:将水池子内的水抽取到实验台,然后借助潜水泵将水中的丙烯腈抽入气相色谱分析仪分析,分析出丙烯腈含量。
3、校准:用校准器将气相色谱仪进行校准,以补偿噪声、漂移等,以便获得精确的分析结果。
4、结果:从气相色谱分析仪得出的结果中,可以获得丙烯腈含量。
通过气相色谱法可以快速而准确地测定水中的丙烯腈含量,具有很高的效率和准确性,是一种可靠的分析方法。
1 适用范围1.1 本标准适用于固定污染源有组织排放和无组织排放的丙烯腈测定。
1.2 当采样体积为30L时,方法的检出限为0.2 mg/m3。
方法的定量测定浓度范围为0.26~33.0 mg/m3。
2 方法原理丙烯腈(CH2=CHCH2CN)用活性炭常温吸附富集,再经二硫化碳常温解吸,解吸液中各组分通过色谱柱得到分离后进人氢火焰离子化检测器(FID),从测得的丙烯腈色谱峰高(或面积),对解吸液中丙烯腈浓度定量,最后由解吸液体积、浓度和采样体积计算出气体样品中丙烯腈的浓度。
3 引用标准下列标准所包含的条文,通过在本标准中引用而构成为本标准的条文:GB 16297-1996 大气污染物综合排放标准GB 16157-1996 固定污染源排气中颗粒物测定和气态污染物采样方法4 试剂和材料4.1 丙烯腈:色谱纯(或分析纯,但必须对丙烯腈无色谱干扰峰)。
4.2 二硫化碳:分析纯(对丙烯腈的色谱测定无干扰峰,否则需进行蒸馏,取46~47℃的馏分)。
4.3 气相色谱固定相:GDX-502, 60~80目。
4.4 氮气:纯度99.99%,并用分子筛或活性氧化铝净化.4.5 氢气:纯度99.99%,并用分子筛或活性氧化铝净化。
4.6 空气。
4.7 活性炭吸附管活性炭吸附管的结构如图1所示。
玻璃管的两端熔封密闭,并配有两个塑胶帽盖,以备采样完毕后盖紧密闭用。
管内填装活性炭粒度为20~40目,A段含100mg, B段含50mg。
A段活性炭前的玻璃棉上压着一个V字型弹簧钩,以免炭粒松动。
活性炭应对气态丙烯腈有很强的吸附能力,并可用二硫化碳解吸被吸附的丙烯腈。
目前市售的用于采集空气中有机蒸气,并以二硫化碳作解吸溶剂的活性炭吸附管能满足要求。
4.8 丙烯腊标准储备液:c=10. 0 mg/ml。
用分析天平准确称取一定t的丙烯腈(4.1)于容量瓶中,小心加人二硫化碳至刻度,配制成溶液的丙烯腈浓度为10.0 mg/ml,作为储备液,密闭存放于低温(4~8℃)下,备用。
存放期不得超过一个月。
4.9 丙烯腈标准使用液:c=1.00 mg/ml。
取1.00ml丙烯睛标准储备液(4.8)于10ml容量瓶中,用二硫化碳稀释至刻度。
5 仪器5.1 气相色谱仪:附氢火焰离子化检测器。
5.2 色谱柱5.2.1 柱材料:玻璃或聚四氟乙烯。
5.2.2 柱长:3m。
5.2.3 柱内径:3mm。
5.2.4 柱类型:填充柱。
5.2.5 柱内填充物:GDx-502, 60~80目。
5.2.6 色谱柱制备和老化〔参见附录B)5.3 微量注射器:1.0μl。
5.4 采样仪器参考GB 16157-1996中9. 3配里采样仪器。
5.4.1 有组织排放监测采样仪器5.4.1.1 采样管采用不锈钢、硬质玻璃或聚四氟乙烯材质,有适当尺寸,并附有可加温至120℃以上的保温夹套。
5.4.1.2 样品收集装置活性炭吸附管(4.7)。
5.4.1.3 流量计量装置见GB 16157-1996中9.3.65.4.1.4 抽气泵见GB 16157-1996中9.3.7。
5.4.1.5 连接管聚四氟乙烯软管或内衬聚四氟乙烯薄膜的硅橡胶管。
5.4.2 无组织排放监测采样仪器5.4.2.1 引气管聚四氟乙烯软管,头部接一玻璃漏斗.5.4.2.2 样品收集装置活性炭吸附管(4.7)。
5.4.2.3 流盘计量装置和抽气泵参考GB 15157-1996中9.3.6 和9.3.7配置。
5.4.2.4 连接管同5.4.1.5。
6 样品采集和保存6.1 有组织排放样品采集6.1.1 采样位置和采样点按GB 16157-1996中9.1.1.和9.1.2确定采样位里和采样点。
6.1.2 采样装置的连接参考GB 16157-1996中9.3图28,按采样管、样品收集装置(吸附管)、流量计量装置和抽气泵的顺序连接好采样系统,注意吸附管进气口应垂直向上(见图2),按GB 16157-1996中9.4的要求检查采样系统的气密性和可靠性。
连接采样系统的连接管要尽可能短。
6.1.3 样品采集在采样管口塞适量玻璃棉,然后将其伸人至排气筒内的采样点位置,启动抽气泵,先使排气筒内的气体由旁路吸附管流通,以充分洗涤采样管路,然后再使排气通过吸附管,记录采样时间、温度和流量,采样完毕后应立即取下吸附管,用塑料帽盖将两端盖紧,带回实验室分析。
6.1.4 采样管加热温度控制采样管加热温度应以水气和样品不在采样管璧凝结为原则,但加热温度最高不得超过160℃。
若排气温度接近常温,采样管也不必加热。
6.1.5 采样流量采样流量一般应控制在0.3~1.0 L/min之间。
当温度高于30℃时,采样流速应降低,不要超过0.5 L/min,以保证B段活性炭吸附量小于吸附总量的2%。
6.1.6 采样量对每支活性炭吸附管,采样童应控制在二硫化碳解吸液中丙烯腈浓度为10~400μg/ml。
每支活性炭吸附管的最大采样量一般不超过1.6mg,且B段活性炭吸附的丙烯腈应不超过被吸附丙烯腈总量的2%。
6.2 无组织排放样品采集6.2.1 采样位置和采样点按GB 16297-1996中附录C的规定确定无组织排放监控点的位置,或按其他特定的要求确定采样点的位置。
6.2.2 采样装置的连接按引气管(如无必要,可不接引气管)、吸附管、流量计量装置、抽气泵的顺序连接采样系统(参看6.1.2)。
6.2.3 样品采集、采样流量和采样量等均参照6.1相应部分。
6.3 样品保存采样后的活性炭管,在两端塞紧帽盖的情况下避光保存,低温下(8℃以下)保存最多不超过7天。
7 分析步骤7.1 色谱条件7.1.1 层析室温度:130℃。
7.1.2 进样器温度:150℃。
7.1.3 检测器温度:150℃。
7.1.4 氮气(4.4)流速:30 ml/min。
1.1.5 氢气(4.5)流速:40 ml/min。
7.1.6 空气(4.6)流速:350 ml/min。
7.2 校准曲线绘制取不同体积丙烯腈标准使用液(4. 9)于6个10 ml容量瓶中,分别加入二硫化碳稀释至刻度,配制成浓度为10.0~500μg/ml标准系列溶液(见表1)摇匀待测。
表1 丙烯腈标准系列溶液编号 1 2 3 4 5 6 标准使用液体积,ml 0.10 0.50 1.00 2.00 3.00 5.00 二硫化碳体积,ml 9.90 9.50 9.00 8.00 7.00 5.00 丙烯腈浓度,μg/ml 10.0 50.0 100 200 300 500按7.1规定的气相色谱操作条件,分别取上述标准系列溶液注人气相色谱仪,进样量为 1.0μl,测定丙烯腈峰高(或峰面积)。
每个浓度的溶液重复进样三次,取平均值,以浓度ci (μg/ml)与对应峰高hi(或面积Ai)绘制校准曲线,并计算得到校准曲线的线性回归方程。
7.3 样品测定7.3.1 样品的解吸将采集样品后的活性炭吸附管两端的帽盖打开,用一根带钩的铁丝取出压在A段活性炭粒上的V字型弹簧钩,将A段活性炭(包括上端玻璃棉)和B段活性炭(包括海绵)分别转移到两支干燥的具磨口的试管内,并立即用吸管移取2.00ml和1.00ml二硫化碳,分别加人盛A段和B段活性炭的试管内,迅速盖紧试管塞,不断轻轻振摇试管,使二硫化碳能和活性炭充分接触混和,30min后进行气相色谱测定。
7.3.2 气相色谱测定按绘制校准曲线相同的气相色谱工作条件调节仪器,对7.3.1中得到A 段和B段活性炭的二硫化碳解吸液分别作气相色谱测定。
取解吸液的上层清液进样,每次进样量为1.0μl,重复进样三次,取丙烯腈色谱峰高(或面积)的平均值定量。
8 计算和结果表示8.1 定性分析以丙烯腈标样的色谱峰保留时间定性。
标准色谱图参见图3。
首次分析成分复杂的样品,且对定性结果有疑间时,应采用双柱定性。
辅助定性的柱特征和色谱条件详见附录A。
若经双柱定性后,对定性结果仍有疑间时,可采用GC/MS等其他方法和手段进一步定性。
8.2 定量分析由测得的丙烯腈色谱峰高(或峰面积)平均值,直接在校准曲线上查得丙烯腈浓度ca 和cb,再根据采气体积计算气体样品中的丙烯腈浓度。
计算式如下:式中:cR——采气样品中丙烯腈浓度,mg/m3 ;ca——A段活性炭解吸液中丙烯腈浓度,μg/ml;cb——B段活性炭解吸液中丙烯腈浓度,μg/ml;Va——A段活性炭的二破化碳解吸液体积,ml;Vb——B段活性炭的二琉化碳解吸掖体积,ml;Vnd——换算到标准状态下的干采气体积,L。
(按GB 16157-1996中10.1或10.2计算)。
8.3 丙烯腈有组织排放的“排放浓度”计算按GB 16157-1996中11.1.2计算丙烯腈的“排放浓度”。
8.4 丙烯腈有组织排放的“排放速率((kg/h)”计算按GB 16157-1996中11.4计算丙烯腈的“排放速率”.8.5 丙烯腈的“无组织排放监控浓度值”计算8.5.1 一个监控点的丙烯腈平均浓度计算按下式计算一个无组织排放监控点的丙烯腈平均浓度。
式中:c——一个无组织排放监控点的丙烯腈平均浓度;ci——一个样品的丙烯腈浓度;n——一个无组织排放监控点采集的样品数。
8.5.2 “无组织排放监控浓度值”的计算按GB 162971996附录C中C2.3计丙烯腈的“无组织排放监控浓度值”。
9 精密度和准确度9.1 精密度浓度为14.1 mg/m3和32.2 mg/m3的丙烯腈标准样品,经五个实验室分别用浓度为14.1 mg/m3和3Z.2 mg/m3的丙烯腈标准样品。
经五个实验室测定,得到方法的平均相对误差分别为 2.0%(范围在0~3.6%之间)和 5.6%(范围3.7%~7.8%之间)。
10 说明10.1 本方法中甲醇、乙醛、丙烯醛、乙腈、丙腈等一般不干扰测定。
若遇干扰物质量稍大而影响丙烯腈出峰时,可将色谱柱内填充剂60~80目的GDX-502改为80~100目,则可得到更好的分离效果。
10.2 二硫化碳沸点较低,极易挥发,因此在配制标准溶液和对样品进行解吸时,均应注意随时盖紧容器的磨口塞。
室内温度不要超过30℃,否则应将盛解吸液的试管置于冷水中。
10.3 二硫化碳不溶于水,使用过的玻璃仪器可用热的稀碱溶液清洗,例如,用5%Na2CO3溶液加热后作洗涤剂,然后用清水冲洗干净后,干燥备用。
丙烯腈辅助定性色谱柱条件A1 固定相β,β一氧二丙腈:6201(60~80)目釉化)二20:80 A2 色谱柱特征A2.1 柱长:2m。
A2.2 柱内径:4m。
A2.3 柱材质:不锈钢。
A2.4 检测器:氢火焰离子化(FID)。
A3 色谱条件A3.1 柱温:80±0.5℃。
A3.2 氮气流速:80ml/min。
A3.3 氢气流速:50ml/min。
A3.4 空气流速:500ml/min。