文件变更履历表
- 格式:xls
- 大小:22.50 KB
- 文档页数:2



供应商管理程序编号:CG-QP-02版本:A/0起草部门:采购中心编制:审核:批准:2022年09月01日发布 2022年09月01日实施本文件适用于******集团及其下属子公司1 目的为保证公司选择、评估及控制供应商的客观性、公正性、科学性,确保外部提供的过程、产品和服务满足要求。
2 范围本程序适用于产品原辅材料的供应商、物流服务方、检定或校准机构、试验测试机构等供应商的管理。
3 定义3.1 供应商提供产品或服务的个人或组织。
供应商可能是产品/服务或信息的生产商、经销商、零售商或供应商。
4 职责4.1 采购中心负责供应商的寻找、组织评估、稽核、定期主导评审、淘汰。
负责合格供方的评价;对供方供货交期、价格、服务状况进行评价。
4.2 品质中心负责供应商质量保证能力的监督、辅导;对供方供货质量状况进评价;并根据此评价向采购中心提出暂停或解除其合格供方资格的建议。
4.3 其他中心及部门负责协助进行供应商评审、辅导、业绩统计提供及评价。
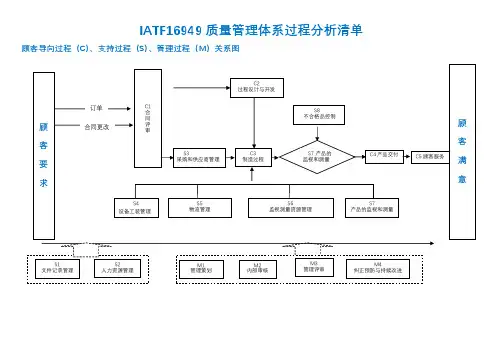
5 流程图供应商策划供应商调查N供应商评价Y供应商选择供应商绩效评定供应商连续评价供应商质量体系开发供应商质量体系审核Y合格供方撤销N资料归档6 工作程序6.1 供应商策划6.1.1 采购中心负责确定公司采购的产品,研发中心负责策划规定采购产品的技术要求,采购产品主要指用于生产和组成产品的材料,有时也包括适当的外包服务等,以下称采购材料。
6.1.2公司根据所采购材料对产品质量的影响程度,将采购的材料分为关键材料A、重要材料B、一般材料C三个级别,不同级别实行不同的控制要求。
其中关键材料如生产用原料(钣金件、塑胶外壳、电器件、机加工件等),重要材料如标准件、油漆、焊接材料、包装材料等,一般材料如包装材料、检测服务、物流服务、办公用品等。
6.1.3 根据相应的材料类别,将供应商分为A、B、C三类。
6.2 供应商调查6.2.1 由采购中心视公司实际需求寻找适合的供应商,同时收集如质量、服务、交期、价格等方面资料作为筛选的依据。

制订部门品保部公布日期页次5/10
5.4.6 校验之仪器与工具由实验室提供书面之校验记录,内校记录于
「仪器与工具内校报告」中(外校由外部实验室提供报告),
同时依据【仪器与工具校验指导书】之允收标准判定,并在仪
器与工具上贴上适当校验标签:
(1)免校标签(绿色):
(2)校验合格标签(绿色):
(3)限制使用标签(橙色):
(4)停止使用标签(红色):
5.4.7 当校验完成后,应在「仪器及工具履历卡」中记录校验信息。
5.5 不良品追溯:
5.5.1 仪器及工具于有效期限内不合格时,产品之追溯:品保人员确
认仪器及工具不合格时,应以质量异常联络单发至仪器及工具
使用单位主管,由生产单位主管协同品保单位各工站QC展开追
踪。
一目的。
为确保新产品顺利导入量产阶段,能提供正确完整的技术文件资料及验证新产品的成熟度,以顺利大量生产。
二组织与权责。
1 研发:对策分析与设计变更,提供样品及技术相关文件资料及零件采购资料。
2 工程:(1) NPI工程师对接研发学习新产品技术,产品特性进行生产作业评估。
(2) NPI工程师安排试产时程表及召开工程试产检讨会,工程问题分析,对策导入。
(3) IE工程师制程安排,包括生产线的评估,绘制SOP,QC工程图。
(4) ME工程师负责治具的设计准备,制程管制,机器设备架设,参数设定及问题分析等。
(5) ME工程师规划新产品之测试策略,测试设备,治具及软体。
负责生产线测试设备的架设,提供测试SOP,测试计划及测试产出分析。
(6) PE工程师跟线确认制程是否与SOP QC工程图相符,指引操作,不良分析统计,异常处理跟进。
3 品质。
(1)产品设计验证测试(Design Verification:DVT)。
(2)功能及可靠度确认。
(3)负责再次确认PVT和DVT的结果是否符合工程规格及客户规格。
(PVT全称为Process Verification Test,意为小批量过程验证测试,硬件测试的一种,主要验证新机型的各功能实现状况并进行稳定性及可靠性测试.DVT是Design Verification Test的简称,设计验证测试,是硬件生产中不可缺少的一个检测环节,包括模具测试、电子性能、外观测试等等。
)(4)试产原材料的确认。
4 资材:(1)采购负责注塑五金 PCB线材等零部件委托加工及材料采购。
(2) PMC负责人机料法环的沟通准备安排或者插入新产线试产。
(3)仓储根据实际需求发料/跟进新物料5 生产:(1)班组长安排新线配合试产。
(2)员工的常规培训指导。
6 文管中心:DVT资料接收确认与管制。
三定义与解释1 NPI:( New Product Introduction) 新产品导入2 BOM: (Bill Of Material)材料清单3 工程试运行(Engineering Pilot Run:EPR):为确认新产品开发设计成熟度所作的试运行与测试。


文件名称:文件编码:编制人及日期:审核人及日期:批准人及日期:发放部门:质量保证部生效日期: 2022 年 12 月 01 日发放范围:质量保证部、销售部、办公室建立药品不良反应数据采集与处理操作规程,加强药品不良反应信息的采集和处理能力。
合用于公司药品不良反应相关数据的采集和处理。
药品不良反应监测与报告专职人员、其他药品安全委员会成员。
1. 药品不良反应数据的采集1.1 医疗机构1.1.1 医药代表采集途径指定医药代表,通过日常拜访医生、药师或者通过电子邮件、电话、传真等方式沟通,采集临床发生的药品不良反应信息。
1.1.2 利用经销商与医疗机构合同采集经销商在与医疗机构签订药品购销合同时,让医疗机构充分知晓不良反应报告的责任,鼓励医生向公司报告不良反应。
1.2 药品经营企业1.2.1 直接从药品零售企业采集公司销售人员直接从药品零售企业采集不良反应信息,通过日常拜访、电话等方式沟通并采集1.2.2 通过药品经销商采集不良反应在与经销商签订药品购销合同或者销售协议时,明确经销商的相关责任,敦促其采集不良反应。
1.3 电话和投诉1.3.1 药品说明书、标签中发布的电话的监管通过销售部、办公室及质量部电话的接听,记录并采集相关的不良反应信息。
1.3.2 通过法律诉讼渠道获悉的不良反应的需报告1.4 学术文献学术文献是高质量的药品不良反应信息来源之一,应定期对文献进行检索,并报告文献中涉及的不良反应。
1.4.1 频率:文献检索频率原则上每一个月进行一次1.4.2 检索库:中国知网(CNKI)、维普网(VIP)、万方数据库等国内文献数据库和PubMed、Embase、Ovid等国外文献数据库。
要求至少要同时检索两个数据库。
1.4.3 文献类型个案报导、病例系列、不良反应综述等,此外临床有效性和安全性研究、荟萃分析等文献来源也可能涉及到不良反应。
个案报导(对单个患者的不良反应进行描述和讨论)病例系列(对多个患者同一性质的不良反应进行描述及讨论)1.5 互联网及相关途径利用公司门户网站采集,在公司网站发布完整的产品说明书并及时更新,提供药品不良反应相关知识和合理用药指导。