欧盟医疗器械指令MDD 试卷答案
- 格式:doc
- 大小:44.00 KB
- 文档页数:2

欧洲共同体理事会关于医疗器械的93/42/EEC指令1993年6月14日欧洲共同体理事会考虑到建立欧洲经济共同体的条约,特别是其第100a条;考虑到欧洲共同体委员会提交的议案;考虑到与欧洲议会合作;考虑到经济与社会委员会的意见;鉴于应在欧洲共同体内部市场范围内正式通过必要的措施;鉴于欧洲共同体内部市场是一个确保商品、人员、服务和资本自由流通的无内部边界的区域;鉴于各成员国有关医疗器械的安全、健康保护和工作特性的法律、法规和行政条款的内容和范围是不同的;鉴于各成员国之间对这类器械的认证和检验程序存在差异;鉴于这种差异在欧洲共同体内形成贸易壁垒;鉴于各国针对医疗器械的使用所制定的有关患者、使用者及其他人员的安全和健康保护的条款应予以协调,以保证此类器械在欧洲共同体内部市场自由流通;鉴于协调条款必须与成员国为管理直接或间接与这类器械有关的公共健康和医疗保险计划的资金筹措而采取的措施相区别;鉴于因而只要遵守欧洲共同体法律,这些条款并不影响各成员国实施上述措施的能力;鉴于医疗器械应向患者、使用者及第三方提供高水平的保护并达到制造商赋予其的性能水平;鉴于因此保持和提高各成员国已达到的保护水平是本指令的基本目的之一;鉴于在1965年1月26日欧洲共同体理事会关于使有关特许专卖药品的法律、法规或行政措施趋于一致的65/65/EEC指令中,某些医疗器械是用于施药的;鉴于在这种情况下,医疗器械投放市场通常由本指令管理,而药品投放市场由65/65/EEC指令管理;鉴于如果这种器械投放市场的方式使器械与药品构成一种规定只供组合使用且不能再次使用的整体,则这个单一整体产品应由65/65/EEC指令管理;鉴于必须将上述器械与包含某种物质,特别是当其单独使用时,按65/65/EEC指令可视为药物的医疗器械相区别;鉴于在这种情况下,若这种物质能配合医疗器械对人体产生辅助作用,则这类器械投放市场由本指令管理;鉴于这类物质的安全性、质量和有效性必须比照欧洲共同体理事会1975年5月20日关于使成员国有关分析标准、药物毒理学标准和临床标准及特许专卖药品试验协议的法律趋于一致的75/318/EEC指令规定的适当方法加以验证;鉴于本指令附录中的基本要求和其他要求,包括对“减小”或“降低”危险的任何引用,在解释和实施时必须考虑设计时的技术现状和实际做法及高水平的健康和安全相适应的技术与经济条件;鉴于按照1985年5月7日欧洲共同体理事会关于技术协调与标准化新方法的决议所规定的原则,有关医疗器械设计和生产的规定必须限于满足基本要求所必须的条款;鉴于因为这些要求是基本的,因此它们应取代各国相应的条款;鉴于实施基本要求应审慎考虑设计时的技术水平和与高水平的健康及安全保护相适应的技术、经济条件;鉴于1990年6月20日欧洲共同体理事会关于使成员国有关有源植入式医疗器械的法律趋于一致的90/385/EEC指令是新方法指令在医疗器械领域中的首次应用案例;鉴于为了使欧洲共同体的统一规定适用于所有医疗器械,本指令基本上是以90/385/EEC指令的条款为依据的;鉴于为此必须修订90/385/EEC 指令,以便放入本指令规定的一般性条款;鉴于电磁兼容性问题是医疗器械安全的一个组成部分;鉴于就1989年5月3日欧洲共同体理事会关于使成员国有关电磁兼容的法律趋于一致的89/336/EEC指令而言,应包含这方面的专门规定;鉴于本指令应包括有关发射电离辐射的器械在设计和制造方面的要求;鉴于本指令既不影响1980年7月15日欧洲共同体理事会80/836/Euratom指令对有关保护公众和工人免受电离辐射危险的基本安全标准的指令进行修订所要求的授权,也不影响欧洲共同体理事会1984年9月3日对接受医疗检查和治疗的人员的辐射防护规定了基本措施的84/466/Euratom指令的实施;鉴于欧洲共同体理事会1989年6月12日关于采取措施鼓励改善工人工作中的安全和健康的89/391/EEC指令以及有关这一问题的专门指令应继续予以实施;鉴于为了证实符合基本要求并使这种符合得到验证,需要制定欧洲协调标准来防止与医疗器械的设计、制造和包装有关的危险;鉴于这类欧洲协调标准是由非官方机构制定的,应保持其非强制性的地位;鉴于为此欧洲标准化委员会(CEN)和欧洲电工标准化委员会(CENELEC)按照1984年11月13日欧洲共同体委员会与这两个机构之间签署的合作总指导原则,被认可为批准协调标准的主管机构;鉴于在本指令中,协调标准是受欧洲共同体委员会委托,由上述两机构之一,或两个机构共同根据欧洲共同体理事会1983年3月18日关于在技术标准和法规领域提供信息程序的83/189/EEC指令,依照上述总指导原则而批准的技术规范(欧洲标准或协调文件);鉴于对协调标准进行修订,欧洲共同体委员会应得到根据83/189/EEC指令建立的常设委员会的帮助;鉴于应采取的措施必须按欧洲共同体理事会87/373/EEC决定中规定的程序Ⅰ而规定;鉴于在特定领域,欧洲药典专著这类现有的形式应包括在本指令的范围内;鉴于因此有几部欧洲药典专著可视为等同于上述协调标准。

欧洲共同体理事会关于医疗器械的93/42/EEC指令1993年6月14日欧洲共同体理事会考虑到建立欧洲经济共同体的条约,特别是其第100a条;考虑到欧洲共同体委员会提交的议案;考虑到与欧洲议会合作;考虑到经济与社会委员会的意见;鉴于应在欧洲共同体内部市场范围内正式通过必要的措施;鉴于欧洲共同体内部市场是一个确保商品、人员、服务和资本自由流通的无内部边界的区域;鉴于各成员国有关医疗器械的安全、健康保护和工作特性的法律、法规和行政条款的内容和范围是不同的;鉴于各成员国之间对这类器械的认证和检验程序存在差异;鉴于这种差异在欧洲共同体内形成贸易壁垒;鉴于各国针对医疗器械的使用所制定的有关患者、使用者及其他人员的安全和健康保护的条款应予以协调,以保证此类器械在欧洲共同体内部市场自由流通;鉴于协调条款必须与成员国为管理直接或间接与这类器械有关的公共健康和医疗保险计划的资金筹措而采取的措施相区别;鉴于因而只要遵守欧洲共同体法律,这些条款并不影响各成员国实施上述措施的能力;鉴于医疗器械应向患者、使用者及第三方提供高水平的保护并达到制造商赋予其的性能水平;鉴于因此保持和提高各成员国已达到的保护水平是本指令的基本目的之一;鉴于在1965年1月26日欧洲共同体理事会关于使有关特许专卖药品的法律、法规或行政措施趋于一致的65/65/EEC指令中,某些医疗器械是用于施药的;鉴于在这种情况下,医疗器械投放市场通常由本指令管理,而药品投放市场由65/65/EEC指令管理;鉴于如果这种器械投放市场的方式使器械与药品构成一种规定只供组合使用且不能再次使用的整体,则这个单一整体产品应由65/65/EEC指令管理;鉴于必须将上述器械与包含某种物质,特别是当其单独使用时,按65/65/EEC指令可视为药物的医疗器械相区别;鉴于在这种情况下,若这种物质能配合医疗器械对人体产生辅助作用,则这类器械投放市场由本指令管理;鉴于这类物质的安全性、质量和有效性必须比照欧洲共同体理事会1975年5月20日关于使成员国有关分析标准、药物毒理学标准和临床标准及特许专卖药品试验协议的法律趋于一致的75/318/EEC指令规定的适当方法加以验证;鉴于本指令附录中的基本要求和其他要求,包括对“减小”或“降低”危险的任何引用,在解释和实施时必须考虑设计时的技术现状和实际做法及高水平的健康和安全相适应的技术与经济条件;鉴于按照1985年5月7日欧洲共同体理事会关于技术协调与标准化新方法的决议所规定的原则,有关医疗器械设计和生产的规定必须限于满足基本要求所必须的条款;鉴于因为这些要求是基本的,因此它们应取代各国相应的条款;鉴于实施基本要求应审慎考虑设计时的技术水平和与高水平的健康及安全保护相适应的技术、经济条件;鉴于1990年6月20日欧洲共同体理事会关于使成员国有关有源植入式医疗器械的法律趋于一致的90/385/EEC指令是新方法指令在医疗器械领域中的首次应用案例;鉴于为了使欧洲共同体的统一规定适用于所有医疗器械,本指令基本上是以90/385/EEC指令的条款为依据的;鉴于为此必须修订90/385/EEC 指令,以便放入本指令规定的一般性条款;鉴于电磁兼容性问题是医疗器械安全的一个组成部分;鉴于就1989年5月3日欧洲共同体理事会关于使成员国有关电磁兼容的法律趋于一致的89/336/EEC指令而言,应包含这方面的专门规定;鉴于本指令应包括有关发射电离辐射的器械在设计和制造方面的要求;鉴于本指令既不影响1980年7月15日欧洲共同体理事会80/836/Euratom指令对有关保护公众和工人免受电离辐射危险的基本安全标准的指令进行修订所要求的授权,也不影响欧洲共同体理事会1984年9月3日对接受医疗检查和治疗的人员的辐射防护规定了基本措施的84/466/Euratom指令的实施;鉴于欧洲共同体理事会1989年6月12日关于采取措施鼓励改善工人工作中的安全和健康的89/391/EEC指令以及有关这一问题的专门指令应继续予以实施;鉴于为了证实符合基本要求并使这种符合得到验证,需要制定欧洲协调标准来防止与医疗器械的设计、制造和包装有关的危险;鉴于这类欧洲协调标准是由非官方机构制定的,应保持其非强制性的地位;鉴于为此欧洲标准化委员会(CEN)和欧洲电工标准化委员会(CENELEC)按照1984年11月13日欧洲共同体委员会与这两个机构之间签署的合作总指导原则,被认可为批准协调标准的主管机构;鉴于在本指令中,协调标准是受欧洲共同体委员会委托,由上述两机构之一,或两个机构共同根据欧洲共同体理事会1983年3月18日关于在技术标准和法规领域提供信息程序的83/189/EEC指令,依照上述总指导原则而批准的技术规范(欧洲标准或协调文件);鉴于对协调标准进行修订,欧洲共同体委员会应得到根据83/189/EEC指令建立的常设委员会的帮助;鉴于应采取的措施必须按欧洲共同体理事会87/373/EEC决定中规定的程序Ⅰ而规定;鉴于在特定领域,欧洲药典专著这类现有的形式应包括在本指令的范围内;鉴于因此有几部欧洲药典专著可视为等同于上述协调标准。

医疗器械MDD医疗器械 MDD Medical Devices-general什么是医疗器械?“医疗器械”是指制造商预定用于人体以下目的的任何仪器、装置、器具、材料或其他物无论它们是单独使用还是组合使用,包括为其正常使用所需的软件:——疾病的诊断、预防、监视、治疗或减轻;——损伤或残障的诊断、监视、治疗、减轻或修补;——解剖学或生理过程的探查,替换或变更;——妊娠的控制医疗器械的评估等级:所有进入欧盟市场的产品,企业必须具有表示自我符合声明的CE 标志,以说明产品符合欧盟制定的相关指令。
医疗器械指令(MDD),MDD指令适用于大多数进入欧盟销售的医疗设备。
它根据不同的要求共分为6个等级,供认证机构评估。
- 设计阶段生产阶段I级自我符合声明自我符合声明I级(测量功能)自我符合声明申报机构I级(灭菌)自我符合声明申报机构IIa级自我符合声明申报机构IIb级申报机构申报机构III级申报机构申报机构认证机构的统一评估包括根据指令规定的基本要求评审技术文件、根据标准EN 46001 或 EN/ISO 13485评审质量体系。
由于美国、加拿大和欧洲普遍以ISO 9001, EN 46001或ISO 13485作为质量保证体系的要求,故建议质量保证体系的建立均以这些标准为基础。
医疗器械的风险分析:EN14411、失效模式及后果分析(FMEA);2、失效树分析(FTA);3、上市后的监控(客户投诉情况等);4、临床经验5、根据EN1441风险分析的一些例子;6、器械的预期用途;7、预期与病人和第三者的接触;8、有关在器械中所使用的材料/元件的风险;9、供给患者或来自患者的能量;10、在无菌条件下生产的器械;11、用于改变病人环境的器械;12、说明用器械;13、用于控制其它器械或药品或与其配合使用的器械;14、不需要的能量或物质的输出;15、易受环境影响的器械;16、带有重要消耗品或附件的器械;17、必要的日常维护和校正;18、含有软件的器械;19、货架寿命有限制的器械;20、延迟或长期使用可能造成的影响;21、普通风险;所有的适用项目必须论述包括可能的危险和降低风险的方法医疗器械CE认证基本要求基本要求是MDD的最重要部分,它包括所有的医疗器械通用要求:一、基本要求(总要求)⑴安全性(任何风险与器械提供的益处相比较,必须在可以接受的范围内,故亦称风险分析);⑵风险的可预防性或被消除性,至少应给予警告(报警系统或警戒报警系统);⑶性能符合性(产品的基本要求);⑷器械性能和安全的效期(器械的安全和性能必须在器械的使用寿命内得到保证。

医疗器械指令(Medical Device Directive)93/42/EEC欧共体医疗器械产品安全共同指令欧洲共同体公报,1993年7月12日,NO. L169/1(此法案对欧共体成员国而言,其公布与否属非强制性)1993年6月14日关于医疗器械的第93/42/EEC号理事会指令欧共体理事会,1考虑到建立欧洲经济共同体的(罗马)条约,特别是其第100a条,2考虑到执委会的提案,以及与欧洲议会的合作,3考虑到经济和社会委员会的意见,4鉴于应就内部市场的完成采取一些措施;鉴于内部市场是一无内部疆界的区域,区域内的货品、人员、服务和资金应可自由流通;5鉴于各成员国间现存有关医疗器械的安全,对健康的保护和功能特性方面的法律、法规和行政命令的内容与范围不尽相同;鉴于各成员国之间对此类器械的认证和检验程序也存在差异;鉴于前述的分歧将在共同体内部构成贸易壁垒;6鉴于为了保护患者、使用者以及必要时其他人员的安全与健康,有关医疗器械使用的国家规定应予以协调,以保证此类器械在内部市场能自由流通;7鉴于协调规定必须与各成员国为管理直接或间接与这类器械有关的公共健康和疾病保险计划的资金筹措所采取的措施相区别;鉴于共同体若与上述措施相符,则这些规定并不影响各成员国实施上述措施的能力;8鉴于医疗器械应向患者,使用者及第三方提供高度的保护并达到制造商赋予其的性能水准;鉴于,因此,维持和改进各成员国已达到的保护水平是本指令的基本目标之一;9鉴于在1965年1月26日的理事会第65/65/EEC号关于使有关根据特许专卖医药产品的法律、法规或管理行为所制定的实施规定趋于一致的指令中某些医疗器械是用于使用药品的;鉴于在这种情况下,医疗器械的市场投放通常受本指令管辖,而药品的市场投放则受第65/65/EEC号指令管辖;鉴于若有某种器械投放市场时器械与其它医疗产品构成一整体的组合单元,并以这种组合形式使用且不能二次使用,则该整体单元产品应受第65/65/EEC号指令管辖;鉴于必须将上述器械与和其它物质组合的医疗器械相区别,特别是若这些物质在单独使用时,按第65/65/EEC号指令可视为药物;鉴于在这种情况下,若这种物质是作为器械的辅助物作用于人体,则这类器械的市场投放受本指令管辖;鉴于,这类物质的安全,质量和效用必须由1975年5月20日理事会第75/318/EEC号关于使成员国有关分析标准,药物毒理学标准和临床标准及特许专卖药品检测协议的法律趋于一致的指令中规定的适当方法加以验证;10 鉴于本指令附录中的基本要求和其它要求包括任何涉及“最低”或“降低”危险的内容的阐述和实施必须考虑设计当时的技术与实际情况,并在符合健康和安全高度保护的原则下考虑技术和经济因素;11 鉴于按照1985年5月7日理事会关于技术协调与标准化新方法的决议所规定的原则,有关医疗器械设计和制造的规则必须限制在满足基本要求所必需的条款内;鉴于因为这些要求是最基本的,因此它们应取代相应的国家规定;鉴于基本要求的实施应谨慎考虑设计当时的技术水平,并在符合健康和安全高度保护的原则下考虑技术和经济因素;12 鉴于1990年6月20日理事会第90/385/EEC号关于使成员国有关有源植入性医疗器械的法律趋于一致的指令是新方法指令在医疗器械领域中的首次应用;鉴于为了统一共同体的规则使之适用于所有医疗器械,本指令在很大程度上是以第90/385/EEC号指令的条款为依据;鉴于同样的原因第90/385/EEC号指令必须增加本指令制定的一般条款的部分;13 鉴于电磁兼容性问题已成为医疗器械安全不可缺少的组成部分;鉴于本指令应包含与1989年5月3日理事会第89/336/EEC号关于使成员国有关电磁兼容的法律趋于一致的指令中的内容有关的特别条款;14 鉴于本指令应包括对有关释放致电离辐射的器械的设计和制造的要求;鉴于本指令既不影响1980年7月15日理事会第80/836/Euratom指令对有关制定一般公众和工人避免离子辐射危险的健康保护的基本安全标准指令的修改中要求的授权,也不影响1984年9月3日理事会第84/466/Euratom号规定有关接受医疗检查和治疗的人员的辐射保护的基本措施的指令的适用;鉴于1989年6月12日理事会第89/391/EEC号关于鼓励改善工作场所中工人的安全和健康的措施介绍的指令和同样主题的其它特别指令应持续适用;15 鉴于为了证实符合基本要求并使该符合性得以验证,有必要协调欧洲标准以避免与医疗器械的设计、制造和包装有关的危险;鉴于这类欧洲协调标准是由非官方立法机构制定的,应保持其非强制性的性质;鉴于到目前为止,欧洲标准化委员会(CEN)和欧洲电工标准化委员会(Cenelec),根据1984年11月13日执委会与这两个机构之间签署的合作通则,已被公认为是制定协调标准的职能机构;16 鉴于本指令的协调标准是一种根据执委会的委托,由上述两机构之一,或两个机构共同根据1983年3月18日理事会第83/189/EEC号关于技术标准和法规领域信息传递程序的指令和上述通则的规定而制定的技术规范(欧洲标准或协调文件);鉴于由于协调标准有可能被修改,执委会应得到根据第83/189/EEC 号指令设立的常设委员会的协助;鉴于所采取的措施必须按理事会第87/373/EEC号决议制定的程序Ⅰ予以阐释;鉴于在特殊领域,如列入欧洲药典专著中的内容,应纳入本指令的框架内;鉴于因此,几部欧洲药典专著应视为等同于前述协调标准;17 鉴于在1990年12月13号决议关于用于技术协调指令不同合格评定程序的各阶段模式中,理事会制定了协调合格评定程序;鉴于将这些模式用于医疗器械可使制造商和公告机构在合格评定中的责任根据有关的器械型式予以确定;鉴于对这些模式的细节所作的补充,根据医疗器械必需验证的性质,证明是合理的;18 鉴于为进行合格评定程序,有必要将器械分为四个产品类别;鉴于分类原则是依据器械技术设计和制造中潜在的危险对人体的易损伤性;鉴于在一般情况下,第Ⅰ类器械具有较低的易损伤性,其合格评定程序可由制造商单独完成;鉴于对第Ⅱa类器械公告机构应在生产阶段强制性介入;鉴于第Ⅱb和第Ⅲ类器械具有较高的潜在危险,公告机构必须对器械的设计与制造阶段进行检验;鉴于第Ⅲ类器械属于最关键的器械,它们在投放市场前需预先就其符合性获得明确授权;19 鉴于在器械的符合性由制造商负责评定的情况下,主管当局必须能够,特别是在紧急情况下,联系到在欧共体内的负责器械市场投放的人员,无论是制造商还是其他在欧共体内经制造商授权的人员;20 鉴于按照一般规则,医疗器械应标示CE标志,表明它们符合本指令的条款,使其能在欧共体内自由流通,并按其预定用途投入使用;21 鉴于为对抗艾滋病和根据1989年5月16日通过的关于在欧共体级别的关于艾滋病预防和控制的进一步行动的理事会决议,用于预防HIV病毒感染的医疗器械必须提供高度的保护;鉴于这类产品的设计和制22 鉴于分类规则通常可对医疗器械进行恰当的分类;鉴于考虑到各种器械的不同性质及该领域内的技术进步,采取的步骤应包括授权执委会决定器械的适当分类或重新分类,必要时调整分类规则;鉴于这些问题与健康保护有密切关系,因此这些决议应按照第87/373/EEC号指令规定的第Ⅲa程序进行;23 鉴于证实符合基本要求可能意味着必须由制造商负责完成临床试验;鉴于为了完成临床试验,必须确定保护公众健康和公共秩序的适当方式;24 鉴于在欧共体级别一体化的医疗器械警戒系统可以更有效地完成健康保护和相关的控制;25 鉴于本指令覆盖了1976年7月27日理事会第76/764/EEC号关于使成员国有关临床用汞柱式温度计最高读数的法律趋于一致的指令中提到的医疗器械;鉴于上述指令应予撤销;鉴于同样原因,1984年9月17日理事会第84/539/EEC号关于使成员国有关用于人或兽医的电子医疗器械的法律趋于一致的指令必须修改。

欧盟MDR 医疗器械法规测卷及答案一、单选或多选选择题(5分一题,漏选得2分,选错得0分)。
1. 我司注射器产品将在2021年8月申请CE认证,我司的审核依据将是( )。
A、MDD 93/42/EECB、MDR (EU) 2017/745C、IVDR (EU) 2017/746D、ISO 13485:20162. 下列产品中,属于MDR指令适用范围的是()。
A、新冠病毒检测试剂B、用于治疗皮肤缺陷的玻尿酸C、隐形眼镜D 、用于实时监测心电图的软件3. 非灭菌I类口罩产品已经在欧洲备案,那么备案有效期至()。
A、2021-05-26B、2024-05-26C、2025-05-26D、长期4. 心脏支架的MDD下的CE产品证书有效期到2020-02-26,产品已经在欧洲情况下,那么欧洲经销商可以销售产品(市场提供)至(),医疗机构可以投入使用到()。
A、2021-05-26B、2024-05-26C、2025-05-26D、长期5. Basic UDI-DI主要用于()。
A、产品规格标签B、符合性声明C、证书D、安全性和临床性能总结6. 在 MDR 规定中,以下附录规定了技术文件的具体内容()。
A、附录 XB、附录 IIC、附录 IIID、附录 IX7. III 类医疗器械需要在()之前,带有UDI编码才可销售。
A、2020-05-26B、2021-05-26C、2022-05-26D、2023-05-268. III类医疗器械的定期安全更新报告“periodic safety update report"(PSUR)需要最少每( )年更新一次,并提供给公告机构审查。
A、1B、2C、3D、不更新9. 一个III类医疗器械需要进行CE认证,认证途径有()?A、附录 X +附录XI Part AB、附录 X +附录XI Part Bc、附录 IX (Chapter I + II +Ill)D、附录 II +附录III + Article 1910. 欧盟的Eudamed数据库将会包含以下信息()。

欧盟医疗器械指令MDD试卷姓名:岗位:得分:一、是非题:(对“√”错“×”)每题3分共30分1.2007/47/EC 是对93/42/EEC 的第5 次修订。
(√)2.含有致癌、导致基因突变及毒性的可能性物质,必须遵循67/548/EC。
(√)3.说明书应有版本及发行日期。
(√)4.对孕妇、哺乳期女性、婴幼儿等产品应明确使用产品的风险。
(√)5.2007/47/EC软件被明确定义为有源医疗器械,必须证明其有效性。
(√)6.注册责任人的信息,需要保密。
(×)7.医疗产品分类,将根据主要作用方式来确定,而不是预期用途。
(√)8.所有器械都将需要临床数据,包括Ⅰ类产品。
(√)9.临床评价可以做对比的方法,不一定做临床。
(√)10.一个设备停用或移走,被另一个相同的设备所替代,将影响分类。
(√)二、选择题:每题5分,共20分。
1..在英国,违反CE标记的惩罚为:(G )A.扣留,罚没 B.5000英镑的罚款C.3个月的监禁 D.通报欧盟-产品消失E.撤出市场或回收所有在用产品 F.追究刑事责任 G.以上的一种和数种并罚2.2007年9月欧盟正式发布对93/42/EEC更新指令2007/47/EC (D )A.这份新指令是自1993年起,对医疗器械指令的一个重大的修改B.2010年3月21日为最终执行期C.所有已经申请CE或即将申请认证的客户所提供的技术文件必须完全符合新指令要求。
D.以上全部3.2007/47/ec医疗器械指令( D )A.2007/47/ec 是新的医疗器械指令。
B.2007/47/ec中仅仅包含3个指令中被修订部分的局部内容,而不是3个指令的完整内容。
C.2007/47/ec 并非新的医疗器械指令。
D. B+C4.2007/47/EC中明确规定记录保存时间不得小于产品使用周期( D )A.自生产之日起五年以备检查B.对植入设备产品生产之日起15年C.拿到证书后,必须到销售国注册,包括Ⅰ类产品D.以上全部三、填充:每空格3分,共30分1.MDD 93/42/EEC共包括23个条款和12附件。
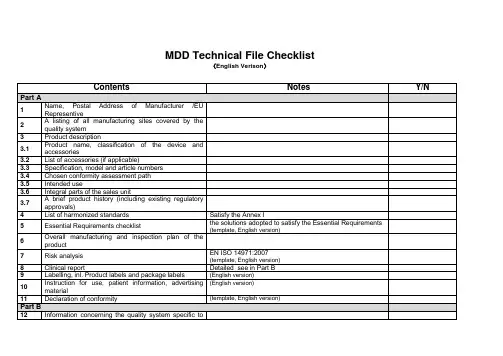


医疗器械指令mdd医疗器械指令MDD是目前欧洲可见到的最为全面的医疗器械方面的规定,在该指令中,共有23个条款和12个附录。
其重要部分包括在以下条款中:第1条款:本指令适用于医疗器械及其附件第2条款:成员国必须确保投放其市场和使用的医疗器械是安全的。
第3条款:所谓“安全”的器械应满足附录1中的基本要求。
第4条款:带有CE标志的医疗器械可在欧盟自由流通。
特殊条款(附录Ⅷ和X)允许使用无CE标志客户定制产品及临床研究的产品。
第5条款:符合协调标准的医疗器械被认为满足基本要求。
第8条款:如发现某种器械不安全,本条款允许成员国采取行动。
第9条款:符合性评价程序依据产品的类型而定,分类规则列于附录Ⅸ。
第11条款:医疗器械必须经过一定的程序(程序Ⅱ-Ⅶ中描述)以证明其满足基本要求。
第17条款:满足基本要求并已通过相应的符合性评价程序的医疗器械必须带CE标志。
医疗器械指令的要求可概括如下:①所有的医疗器械应满足指令的基本要求。
②每种医疗器械在投放市场之前,应通过符合评价程序。
③所有已进行相应的符合性评价的医疗器械应带有CE标志。
满足这些要求的责任在于制造商。
在这里所谓“制造商”指的是把医疗器械以自己的名义的投放市场的人,而不管他是否实际生产、由别人代其生产或仅仅销售该器械,制造商的定义使那些原来仅销售而不生产的公司面临了一种全新的情况。
根据指令,现在他们作为制造商必须满足指令中规定的适用他们的所有的义务。
1分类医疗器械的范围从橡皮膏到心肺机,十分广泛,很难有一个统一的规则适用于所有的医疗器械。
因此医疗器械指令采用了一个分类体系。
它把医疗器械分为四类。
即Ⅰ、Ⅱa、Ⅱb和Ⅲ。
I类产品中还包括无菌或具有测量功能的医疗器械的特例,一般用I*表示。
分类是依据创伤性、使用时间、使用部位以及有无能量等准则进行的。
在医疗器械指令附录Ⅸ****有十八条分类标准规则可作为某种器械的分类指导。
该体系的优点在于它的“灵活性”。
对于新型或很少使用的医疗器械也能确认其类型,而不必列出一个貌似完整却需要经常做修改的医疗器械分类目录。

医疗器械指令MDD一、医疗器械指令MDD1993年6月14日关于医疗器械的理事会指令93/42 / EEC,医疗器械指令旨在确保社区内货物的自由流动,同时为患者,用户和第三方提供高水平的保护,并达到制造商对医疗器械的性能水平。
医疗器械指令定义哪些产品属于其应用领域,它提供了其所涵盖的医疗器械和配件必须遵守的基本要求,并概述了制造商必须应用的合格评定程序,以确保符合必要条件,要求。
二、哪些产品符合医疗器械指令医疗器械93/42 / EEC指令而言,以下定义适用:1.“医疗器械”是指任何仪器,仪器,器具,材料或其他物品,无论是单独使用还是组合使用,包括制造商为了人类而使用的正确应用所必需的软件:2.诊断,预防,监测,治疗或缓解疾病;3.调查,更换或修改解剖学或生理过程;4.以及未通过药理学,免疫学或代谢手段在人体内或人体上实现其主要预期作用,但可通过此类手段协助其作用;5.“附件”是指虽然不是设备,但其制造商专门用于与设备一起使用以使其能够根据设备制造商所预期的设备的使用而使用的物品;三、“体外诊断医疗设备”是指制造商打算用于体外的任何医疗设备,其是试剂,试剂产品,校准器,控制材料,试剂盒,仪器,仪器,设备或系统,无论是单独使用还是组合使用。
用于检查来自人体的标本,包括血液和组织捐赠,仅用于或主要用于提供信息:1.关于生理或病理状态,关于先天性异常,或确定与潜在接受者的安全性和相容性,或监测治疗措施。
2.标本容器被认为是体外诊断医疗设备,标本容器”是指那些真空型或非真空型装置,它们的制造商专门用于主要容器和保存。
3.定制设备”是指根据合格医师的书面处方特别制造的任何设备,该处方在其职责范围内赋予特定的设计特征,并且仅供特定患者使用,批量生产的设备需要进行调整以满足医生或任何其他专业用户的特定要求,不被视为定制设备;4.用于临床研究的装置”是指在适当的人体临床环境中进行附件X第2.1节所述的调查时,适合合格医师使用的任何装置。

医疗器材产品安全指令93/42/EEC欧体医疗器材产品安全共同指令之内容欧洲共同体公报,1993年7月12日,NO.L169/1(此法案对欧体会员国而言,其公布与否非属强制性)1993年6月14日理事会第93/42/EEC号指令关于医疗器材欧洲共同体理事会依据欧洲经济体所制订的罗马条约,特别是第100a条规定,依据执委会的建议案(1)配合欧洲议会(2),依据经济暨社会委员会的意见(3)鉴于内部市场之完成应采取一些措施;鉴于内部市场是一无内部疆界之区域,区域内之货品,人员,服务及资金应可自由流通;鉴于各会员国间现存有关医疗器材之安全,对健康之保护及使用特性方面之法律,法规及行政命令之内容与范围不尽相同;鉴于各会员国对此器材之验证及检验程序也不相同;鉴于前述之分歧将阻碍共同体内的贸易活动;鉴于医疗器械之使用对病患,使用者,甚至其他人有关安全及健康保护的相关国家规定应加以调和,以保证此类器材在内部市场能自由流通;鉴于调和之规定必然与各会员国采取之部分措施有所不同,这些措施是为筹措公共健康与疾病保险计划之基金,且直接或间接与医疗器材有关;鉴于共同体若与上述措施相符,则这些规定并不影响会员国落实上述措施的能力;鉴于医疗器材应提供病患,使用者及第三者高度之保护,且应该达到厂商所要求之性能水准;鉴于维持或改进各会员国对病患等保护的程度乃本指令目的之一;注 1:欧洲共同体公报,NO.C237,1991年9月12日及NO.C251,1992年9月28日,P.40注 2:欧洲共同体公报,NO.C150,1993年5月31日及NO.C176,1993年6月28日,注 3:欧洲共同体公报,NO.C79,1991年3月30日 P1鉴于部分医疗器材是符合1965年1月26日理事会第65/65/EEC号指令,与专卖医药产品有,鉴于医疗器材之上市基本上由本指令规范,关之法律,法规或管理行为所订之实施规定(4)但医疗产品之上市则受65/65/EEC号指令规范;鉴于若有某种器材须与其他医疗产品组成一完整的产品而上市销售、使用,且无法二次使用时,则该组合产品应受65/65/EEC号指令规范。
欧盟医疗器械法规(eu) 2017-746(英文+中文版)Regulation (EU) 2017/746 on medical devices, also known as the Medical Devices Regulation (MDR), is a comprehensive piece of legislation that sets out the rules and requirements for medical devices in the European Union. The regulation was adopted on April 5, 2017, and will fully apply starting from May 26, 2021. It replaces the previous Medical Devices Directive (MDD) and Active Implantable Medical Devices Directive (AIMDD).The main objective of the MDR is to ensure the safety and performance of medical devices while promoting innovation and access to market for manufacturers. The regulation introduces several new requirements for medical devices, including stricter pre-market assessment procedures, enhanced post-market surveillance obligations, and improved traceability and transparency.One of the key changes introduced by the MDR is the new classification system for medical devices, which is based on potential risk to patients. The regulation also establishes a Unique Device Identification (UDI) system to improve traceability of devices throughout the supply chain. In addition, the MDR requires manufacturers to provide more clinical evidence forhigh-risk devices and to demonstrate compliance with general safety and performance requirements.The MDR also strengthens the role of notified bodies, which are responsible for assessing the conformity of medical devices with the regulatory requirements. Notified bodies must now meet higher standards of competence and independence, and are subject to stricter supervision by national authorities.Overall, the MDR aims to improve the safety and effectiveness of medical devices on the EU market, while also enhancing the transparency and predictability of the regulatory framework. By ensuring that medical devices meet high standards of quality and performance, the regulation ultimately benefits patients, healthcare professionals, and manufacturers alike.。
欧盟医疗器械指令MDD 试卷
姓名:岗位:得分:
一、是非题:(对“√”错“×”)每题3分共30分
1.2007/47/EC 是对93/42/EEC 的第5 次修订。
(√)
2.含有致癌、导致基因突变及毒性的可能性物质,必须遵循67/548/EC。
(√)
3.说明书应有版本及发行日期。
(√)
4.对孕妇、哺乳期女性、婴幼儿等,产品应明确使用产品的风险。
(√)
5.2007/47/EC中,软件被明确定义为有源医疗器械,必须证明其有效性。
(√)
6.注册责任人的信息,需要保密。
(×)
7.医疗产品分类,将根据主要作用方式来确定,而不是预期用途。
(√)
8.所有器械都将需要临床数据,包括Ⅰ类产品。
(√)
9.临床评价可以做对比的方法,不一定做临床。
(√ )
10.一个设备停用或移走,被另一个相同的设备所替代,将影响分类。
(√)
二、选择题:每题5分,共20分。
1..在英国,违反CE标记的惩罚为:(G)
A.扣留,罚没B.5000英镑的罚款C.3个月的监禁D.通报欧盟-产品消失
E.撤出市场或回收所有在用产品F.追究刑事责任G.以上的一种或数种并罚2.2007年9月欧盟正式发布对93/42/EEC更新指令2007/47/EC ( D )
A.这份新指令是自1993年起,对医疗器械指令的一个重大的修改
B.所有已经申请CE或即将申请认证的客户所提供的技术文件必须完全符合新指令要求。
C.2010年3月21日为最终执行期D.以上全部
3.2007/47/EC医疗器械指令( D )
A.2007/47/EC中仅仅包含3个指令中被修订部分的局部内容,而不是3个指令的完整内容。
B.2007/47/EC 是新的医疗器械指令。
C.2007/47/EC 并非新的医疗器械指令。
D.A+C 4.2007/47/EC中明确规定记录保存时间不得小于产品使用周期(D)
A.自生产之日起五年以备检查B.拿到证书后,必须到销售国注册,包括Ⅰ类产品C.对植入设备产品生产之日起15年D.以上全部
三、填充:每空格3分,共30分。
1.MDD 93/42/EEC共包括23 个条款和12 附件。
2.医疗器材指令的附录9将医疗器材分类成18项规则。
制造商需视产品预期用途来做产品分类:
a.规则1 to 4 非侵入式医疗器材
b.规则5 to 8 侵入式医疗器材
c.规则9 to 12 主动医疗器材的进一步规划
d.规则13 to 18 特殊规则
3.“CE”标志是一种安全认证标志,被视为制造商打开并进入欧洲市场的护照。
a.凡是贴有“CE”标志的产品就可在欧盟各成员国内销售,无须符合每个成员国的要求,从而实现了商品在欧盟成员国范围内的自由流通。
b.在欧盟市场“CE”标志属强制性认证标志,这是欧盟法律对产品提出的一种强制性要求,如果产品上没有,将被认为是违法行为。
c.不论是欧盟内部企业生产的产品,还是外部其他国家生产的产品,要想在欧盟市场上自由流通,就必须加贴“CE”标志。
d.指令要求没有CE标志的产品,不得上市销售,已加贴CE标志进入市场的产品,发现不符合安全要求的,应责令从市场收回,将被限制或禁止进入欧盟市场或被迫退出市场。
四、简答题:每题10分,共20分。
1.什么是“CE”技术文件(TCF) ?
a.技术文件是合格评定活动的文件化表述形式,证明产品对指令的符合性;
b.该文件由生产厂家负责制定,旨在提供产品有关的设计、生产和使用的信息;
c.文件的内容包括证实产品对于适用要求的符合性证据。
2. 欧盟委员会对医疗器械制定了哪三个指令?并简单例举说明。
三个指令分别是:
1.有源植入性医疗器械指令(AIMD,90/385/EEC),适用于心脏起搏器,可植入的胰岛素泵等有源植入性医疗器械。
AIMD于1993年1月1日生效。
从1995年1月1日强制实施。
2.活体外诊断器械指令(IVD,98/79/EEC)),适用于血细胞计数器,妊娠检测装置等活体外诊断用医疗器械。
该指令于1998年10月27日生效,各成员国最晚不迟于2000年6月7日生效并正式实施。
3.医疗器械指令(MDD,93/42/EEC),适用范围很广,包括除有源植入性和体外诊断器械之外的几乎所有的医疗器械,如无源性医疗器械(敷料、一次性使用产品、接触镜、血袋、导管等);以及有源性医疗器械,如核磁共振仪、超声诊断和治疗仪、输液泵等。
该指令已于1995年1月1日生效,从1998年6月14日起强制执行。