EMEA(欧洲医药品管理局)每月批准新药查询方法
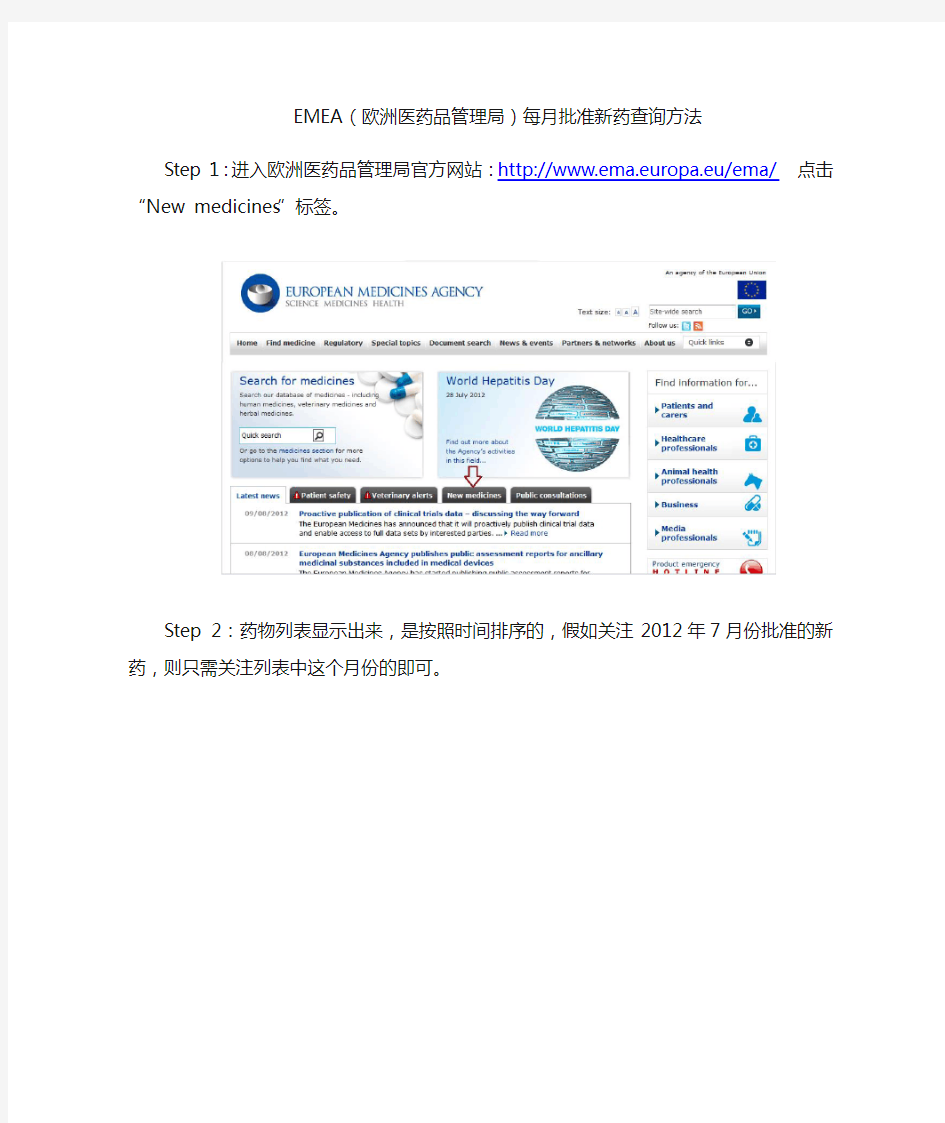
Step 1:进入欧洲医药品管理局官方网站:http://www.ema.europa.eu/ema/点击“New medicines”标签。
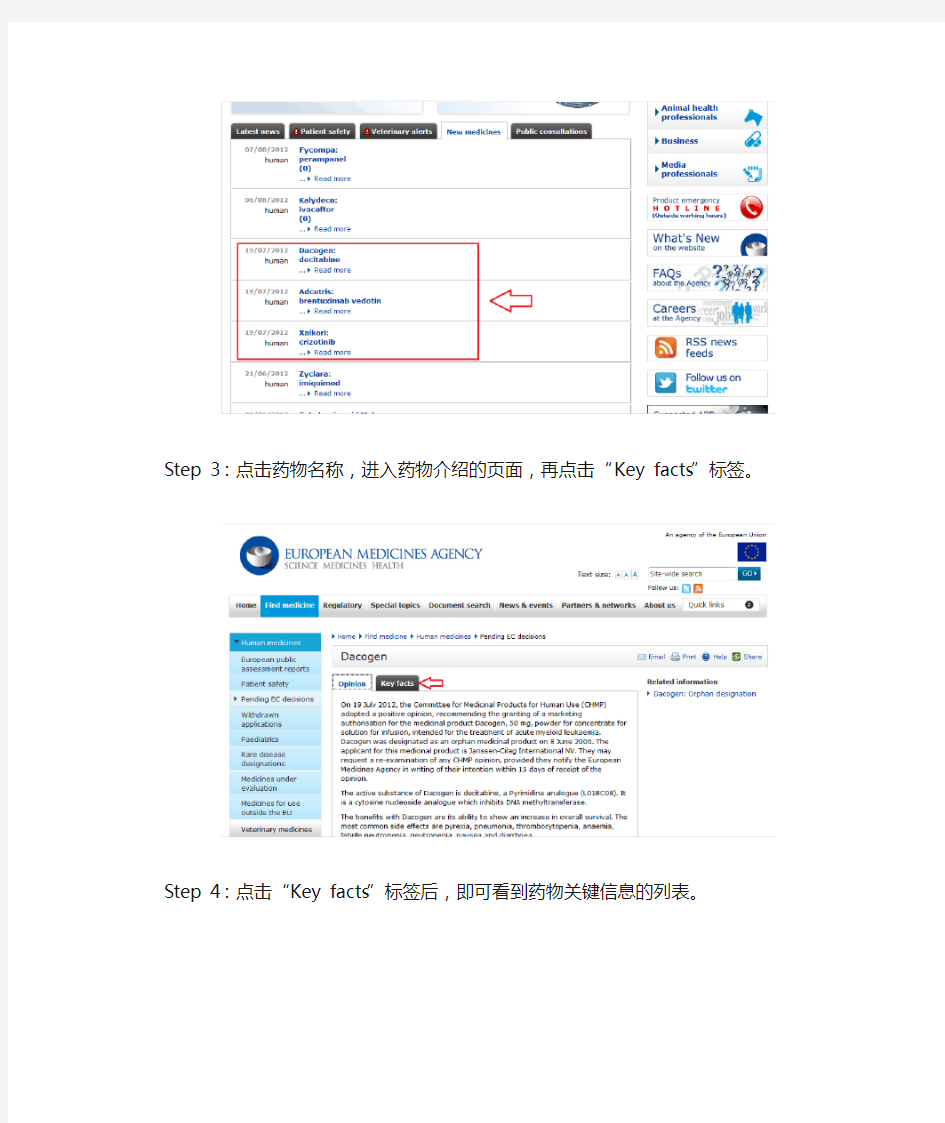
Step 2:药物列表显示出来,是按照时间排序的,假如关注2012年7月份批准的新药,则只需关注列表中这个月份的即可。
Step 3:点击药物名称,进入药物介绍的页面,再点击“Key facts”标签。
Step 4:点击“Key facts”标签后,即可看到药物关键信息的列表。
编号:_________________ 基本药品购销合同范本 甲方:________________________________________________ 乙方:________________________________________________ 签订日期:_________年______月______日
甲方: 乙方: 为构建诚信、公正的药品购销新秩序,维护药品集中招标采购工作成果,提高基本药物配送率,满足临床用药需求,根据《民法典》经双方协商签订如下合同: 一、甲方责任 (一)甲方保证通过河南省医院招标采购网从乙方采购双方协商约定的药品采购目录内的全部药品,非经乙方同意,不得从其他公司采购。 (二)甲方根据用药情况,每月采购计划一般不超过两次,同一品种一般不得超过三个品规。甲方应充分考虑临床应急需要,备足应急药品。 (三)甲方提供建立药品周转库的场所及工作人员,乙方分担基础设施及必备的办公用品费用,每公司五千元(5000元),甲方保证在资金到位后按照药品监管部门要求建立药品周转库。 (四)甲方在合同约定的数量,规格,金额和时限范围内,对乙方配送的合格中标产品,不得以非正当理由拒收或退货。
(五)甲方自收到药品发票之日起最长不得超过30个工作日结算药款。 (六)甲方对乙方配送药品数量、规格、金额、及时性有考评的权利。 二、乙方责任 (一)乙方保证按照在《河南省医疗机构药品集中招标采购挂网目录》下双方协商约定的药品采购目录及本合同涉及的内容,向甲方提供合格的中标药品。 (二)乙方需向甲方药品周转库提供相应的库存药品,以满足药品周转为原则,使甲方能够及时领取到所需药品,库存药品所有权归乙方,在合同履行期内,不得提出库存药品的付款要求。合同终止时库存药品撤回。 (三)乙方应具有满足提供甲方双方协商约定的药品采购目录内全部药品的能力,保证及时足量供应,因市场供应或药品生产企业原因,无法配送的品种,要提前一月通知甲方,停止网上采购,双方协商解决。同一品种不同产地或规格调换的双方签订《xxx公司与xxxxxx卫生院基本药物配送调换备忘》(附件1)已协商约定的药品终止配送的双方签订《xx公司与xxxxxxxx卫生院基本药物配送终止备忘》(附件2)新增加药品配送的双方签订《xx公司与xxxxxxxxxx基本药物配送新增备忘》(附件3) (四)乙方供货的时间和数量以甲方的采购计划或合同为准,急救药品或紧急情况用
现在有三种申报方法 一.申报1.1类药进口(进口药) (1)方法:a.等药品在国外上市后,不在国内生产。申请分包进口(类似于代理)。 b.等药品在国外上市后,完善国内厂房技术,在国内生产。 (2)费用:临床实验37.6万,生产59.39万。 (3)难点:a.需要等药物在国外上市后才可以进行,延长周期。 b.如果在国内生产,需要完善国内厂房技术,延长周期、增加费用。 c.如果国外生产,分包进入国内时可以申请免除临床实验,但是不能确定可以申请成 功。 d.需要所有的临床资料,包括所有的实验数据。 二.申报1.1类新药临床试验(国产药) (1)方法:直接在国内申报1类新药,相关药理毒理研究资料由国外进行,提供相关证明,然后工艺研究、质量研究跟稳定性研究在国内进行,等国内药厂达到生产要求后,将药品在国内生产,按照国内药来报。 (2).费用:申请临床实验19.2万,申请生产43.2万。 (3)难点:a.需要等国内工厂具备相关生产条件后才可以进行申报。 b.无法申请避免临床。 三.申报国际多中心临床试验 (1)基本流程+费用+时间:主要临床基地伦理委员会审(费用...,时间...)+临床审批(费用37.6万,时间205天)+药品清关(费用...+时间...)(时间相对较短) (2)方法:申报程序与国内1.1类化学新药临床试验申报大致相同,但是需要更多的临床资料和
证明性文件。 (3)申请地点:国家药监局北京市西城区宣武门西大街28号大成广场3门一层(总局办公大楼 西侧),邮编100053 (4)流程图: 注:斜线前为一般审批时限,斜线后为特殊审批时限,均为工作日。 (5)申报资料列表: (一)概要部分
药品采购请示文档6篇 Drug purchase request document 编订:JinTai College
药品采购请示文档6篇 小泰温馨提示:请示报告是机关、事业和企业单位在公文往来中经常 用到的重要评议体之一。主要用于向上级机关请求指示、请示批准某 一事项的报告。本文档根据请示报告内容要求展开说明,具有实践指 导意义,便于学习和使用,本文下载后内容可随意修改调整及打印。 本文简要目录如下:【下载该文档后使用Word打开,按住键盘 Ctrl键且鼠标单击目录内容即可跳转到对应篇章】 1、篇章1:药品采购请示文档 2、篇章2:药品采购请示文档 3、篇章3:药品采购请示文档 4、篇章4:申请购买药品的请示范文 5、篇章5:申请购买药品的请示范文 6、篇章6:申请购买药品的请示范文 药品作为一种特殊商品,直接关系到国民生命健康,因此, 药品行业方方面面的变化备受世人关注。下文是药品采购请示,欢迎阅读! 篇章1:药品采购请示文档
为了迎接院运动会、校运会等大型体育赛事到来,我院学生会女生部负责后勤医疗工作。据统计药品不足,尚不能够为运动员提供充足的医疗保障,现需要置办医药用品。按市场价计算,所需药品经费总计300元。故特此申请300元作为本项目购买经费。 以上请示妥否,请批示。 联系人:院学生会XXX部部长XX(xxxxxxx555) 广西XXXXXXXXX学生委员会 XXXX年XX月XX日 篇章2:药品采购请示文档【按住Ctrl键点此返回目录】酒泉市食品药品监督管理局: 我校因教学需要,需购买部分特殊药品(见附表),使用期限为一年。 20XX年12月17日 篇章3:药品采购请示文档【按住Ctrl键点此返回目录】为响应中国石油和化学工业联合会、中国就业培训技术指导中心、中国能源化学工会全国委员会联合举办第六届全国
正好这几天在研究国内的新药研发申请审批流程,一般来说,一种新药(创新药)其大致的流程可以用下图概括: .1
具体来看,各个阶段其耗时也是有差异的:立项(4个月)→临床前研究(9-24个月)→CDE待批临床(大于1年)→临床试验(3-5年)→CDE待批生产(1年-n年)→批文生产转移(约6个月),尤其是重磅新药其从立项到最后获得生产批准文号,直到通过GMP认证上市销售起码要8-10年。拿金赛药业的重磅创新药聚乙二醇重组人生长激素注射液为例,从2005年获批临床,到今年1月获生产 这份流程图和这篇文章给出了药物研发的成功率以及每个步骤所花费的时间。总体来看,每5种进入临床的药物中只有1种能进行所有的试验和批准过程。 新药开发阶段如下: 临床前试验:由制药公司进行的实验室和动物研究,以观察化合物针对目标疾病的生物活性,同时对化合物进行安全性评估。这些试验大概需要3.5年的时间。 研发中新药申请(Investigational New Application, IND):在临床前试验完成后,公司要向FDA提请一份IND,之后才能开始进行药物的人体试验。如果30天内FDA没有发出不予批准的申明,此IND即为有效。提出的IND需包括以下内容:先期的试验结果,后续研究的方式、地点以及研究对象;化合物的化学结构;在体内的作用机制;动物研究中发现的任何毒副作用以及化合物的生产工艺。另外,IND必须得到制度审核部门(the Institutional Review Board)的审核和批准。同时,后续的临床研究需至少每年向FDA提交一份进展报告并得到准许。 .2
临床试验,Ⅰ期:此阶段大概需要1年时间,由20~80例正常健康志愿者参加。这些试验研究了药物的安全性方面,包括安全剂量范围。此阶段的研究同时确定了药物在体内的吸收、分布、代谢和排泄、以及药物的作用持续时间等项目。 临床试验,Ⅱ期:此阶段需要约100到300名志愿患者参与进行一些控制研究,以评价药物的疗效。这个阶段大约需要2年时间。 临床研究,Ⅲ期:此阶段持续约3年时间,通常需要诊所和医院的1000~300名患者参与。医师通过对病患的监测以确定疗效和不良反应。 新药申请(New Drug Application, NDA):通过三个阶段的临床试验,公司将分析所有的试验数据。如果数据能够成功证明药物的安全性和有效性,公司将向FDA提出新药申请。新药申请必须包括公司所掌握的一切相关科学信息。典型的新药申请有10万页甚至更多。根据法律,FDA审核一份NDA的时限应该为6个月。但是几乎所有案例中的新药申请从首次提交到最终获得FDA 批准的过程都超过了这个时限;在1992年对于新分子实体的新药申请平均审核时间为29.9个月。 批准:一旦FDA批准了一份新药申请,此种新药就可以被医师用于处方。公司必须继续向FDA提交阶段性报告,包括所有的不良反应报告和一些质量控制记录。FDA还可能对一些药物要求做进一步的研究(Ⅳ期),以评价药物的长期疗效。 .3
(二)已有国家标准的药品 《药品注册申请表》 1.综述资料 资料编号1、药品名称 资料编号2、证明性文件。 资料编号4、对主要研究结果的总结及评价。 资料编号5、药品说明书样稿、起草说明及最新参考文献。 资料编号6、包装、标签设计样稿。 2.药学研究资料 资料编号7、药学研究资料综述。 资料编号8、药材来源及鉴定依据。 资料编号12、生产工艺的研究资料及文献资料,辅料来源及质量标准。 资料编号15、药品标准草案及起草说明,并提供药品标准物质及有关资料。 资料编号16、样品检验报告书。 资料编号17、药物稳定性研究的试验资料及文献资料。 资料编号18、直接接触药品的包装材料和容器的选择依据及质量标准。 以上申报材料具体要求详见《药品注册管理办法》附件一。 (一)申报资料的一般要求: 1、申报资料按《药品注册管理办法》(国家食品药品监督管理局令第17号)附件一规定的资料顺序编号,按编号分别装订,申报资料首页为申报资料目录。 2、申报资料应使用A4纸打印,内容完整、清楚,不得涂改。 3、资料封面应包含以下信息:药品名称、资料项目编号、项目名称、申请机构联系人姓名、电话、地址,试验资料完成机构名称、主要完成人、参加人、电话、原始资料保存地点。并须加盖各机构公章。 4、资料按套装入档案袋,档案袋封面注明:申请分类、注册分类、药品名称、本袋所属第X套第X袋每套共X袋、原件/复印件、申请机构、联系人、电话。 5、注册申请报送2套完整申请资料(其中至少1套为原件)和1套综述资料(可为复印件),各袋均应包含1份申请表。 6、《药品注册申请表》:从国家食品药品监督管理局网站(https://www.doczj.com/doc/f212687903.html,)下载,按要求填写后打印并保存,用于提交的申请表电子文件与书面申请表的数据核对码必须一致,并一并提交。 7、临床试验总结资料封面应有临床试验组长单位盖章,各临床试验分总结应有试验单位盖章。 (二)申报资料的具体要求: 1、《药品注册申请表》:临床试验完成后申请生产或仅申请新药证书,应重新填写《药品注册申请表》 该表是申请人提出药品注册申请的基本文件,同时也是药监部门对该申请进行审批的依据,其填写必须准确、规范,并符合填表说明的要求。
European Medicines Agency Inspections London, 27 April 2005 EMEA/CVMP/134/02 Rev 2 Consultation CPMP/QWP/227/02 Rev 2 Consultation COMMITTEE FOR MEDICINAL PRODUCTS FOR HUMAN USE (CHMP) COMMITTEE FOR MEDICINAL PRODUCTS FOR VETERINARY USE (CVMP) GUIDELINE ON ACTIVE SUBSTANCE MASTER FILE PROCEDURE DISCUSSION AT THE HMPC November 2005 – January 2006 ADOPTION BY THE HMPC 22 January 2006 DRAFT AGREED BY QUALITY WORKING PARTY February 2006 23 March 2006 ADOPTION BY CHMP FOR RELEASE FOR CONSULTATION 20 April 2006 ADOPTION BY CVMP FOR RELEASE FOR CONSULTATION 30 August 2006 END OF CONSULTATION (DEADLINE FOR COMMENTS) Note: From 1st November 2005, Directive 2004/24/EC1 relating to traditional herbal medicinal products came into force in all Member States in the European Union allowing the establishment of a simplified procedure for the registration of traditional herbal medicinal products for human use. In order to facilitate the use of the ASMF procedure in the area of herbal medicinal products, the Committee for Herbal Medicinal Products proposes an Annex on herbal substances/preparations (see Annex 1, table 3) to the Guideline on the Active Substance Master File procedure. It should be noted that the principles which are outlined in this guideline in relation to traditional herbal medicinal products are equally applicable to other herbal medicinal products, both for Human and Veterinary use, which do not follow the simplified registration procedure. The new table (Annex 1, table 3) takes into account the particularities of herbal substances/preparations whilst also highlighting that this procedure is/can be applied to active substances/preparations of herbal origin, whether they be for human or veterinary use. 1 Directive2004/24/EC of the European Parliament and of the Council of 31 March 2004, amending, as regards traditional herbal medicinal products, Directive 2001/83/EC on the Community code relating to medicinal products for human use.
药品购销合同 甲方(需方):湖南省安仁县中医院 乙方(供方): 为规范我院药品采购行为,保证医院临床用药及时供应,保障人民用药安全有效,根据《合同法》、《药品管理法》等法律法规,经供需双方友好协商达成如下购销协议: 一、供方必须是证照齐全具有合法经营资质的企业,并向需方提供加盖单位红印章的《药品经营企业许可证》复印件、《药品经营质量管理规范认证证书》复印件、《营业执照》复印件、业务员的法人委托书原件、被委托人身份证复印件及其它相关资料等,供需方存档备查。需方应向供方提供加盖单位印章的《医疗机构许可证》复印件。 二、供方只能向需方供应“三证”规定范围内的药品,如供方超范围向需方提供药品,因此造成的一切损失由供方承担。 三、供方要对提供的药品质量负责,不得向需方提供假劣药等不合格药品。供方所提供的药品凡因质量问题所造成的如抽检不合格、药害事件等后果,概由供方承担。如因需方储存和保管不当造成的损失由需方自行负责。 四、供方提供的全部药品均应按标准保护措施进行包装避免污染,需冷藏的药品在运输过程中必须按国家规定使用冷链设备,以防止在转运过程中损坏或变质,确保药品安全无损抵达需方。否则,为此而给需方造成的一切损失由供方承担。 五、需方要求供方遵守以下供药原则:1、不得供应“三无”药品;2、不得供应假冒厂牌和商标及无出厂合格证的药品:3、不得供应即将变质或生产日期较久的药品;4、除双方另有约定外,不得供应有效期少于6个月的药品;5、不得供应不符合包装标志有关规定和储运要求的药品。 六、供方提供进口药品时,须向需方提供加盖供方印章的《进口药品注册证》和《进口药品检验报告书》。 七、需方所造的购药计划必须以农保、医保用药目录为范围,符合我院临床基本用药需要,且经正常审批程序后方可采购。 八、供方须按需方购药计划中规定的药名、规格、数量、产地等要求供应药品,否则需方有权全部或部分拒收。供方接到需方的购药计划后,应在规定时间内将药品送到需方,送货及搬运费由供方承担。未按要求及时交货需方可终止供方送货另选供货公司。 九、需方凭供方药品清单票据(清单内容与实物相符),经需方验收小组验收符合规定要求后方可办理入库手
医院新药采用审批程序 1. 申请 1.1 新药申请需副主任医师以上职称者负责填写《医院新药采用申请表》,科室的正主任签字同意。专科用药需相应的专科申请,中成药一般应有中医科提出申请,西医科室申请中成药须经医院药事管理委员会的中医药专业委员就方解、功能主治等签署意见。 1.2申请表内容包括:药品基本信息、申购理由等。 1.3申请表交药学部药品供应室主任。 1.4药品供应室主任确认表格填写无误、内容完整之后,在表格上标注申请编号和受理日期。2形式审查 2.1药品采购员凭《医院新药采用申请表》对已受理的申请进行形式审查。 2.2药品采购员按《医院新药采用申请表》上记录的药品申请商和销售商的联系方式与其联系,索取资料。 2.2.1一般化学药品应采取下列资料:药品生产企业许可证和经营执照,药品生产批文或试生产批件,新药证书、注册商品批件,物价批文、GMP证书、法定质量标准、药品出厂检验报告书,包装、标签和说明书实样、药品的药学(药学、药理、毒理)资料和临床(临床、不良反应)资料、药品经营企业许可证和营业执照、GSP证书、《药品生产企业质量保证情况调查表》。 2.2.2进口药品还应索取进口药品注册证、中文说明书、进口药品通关单、进口药品检验报告书。 2.2.3进口生物制品还应索取生物制品进口批件或生物制品进口批签发证。 2.2.4麻醉药品、医用毒性药品、精神药品、医用放射性药品应按有关规章制度索取资料。 2.2.5中药成药设计保护品种的还应索取中药保护品种证书。 2.2.6无知识产权纠纷的保证书。 2.2.7以上文件可为复印件,所有文件均须加盖该企业的原印章。 2.3形式审查应关注以下方面 2.3.1证书、批件应注意颁发机关、编号和有效期。 2.3.2国家定价品种应有发改委文件,企业自定价品种应有所在地的物价备案文件。 2.3.3GMP证书应注意其批准内容(全厂认证或生产线认证)。 2.3.4质量标准为国家标准或地标升国标。 2.3.5检验报告书是否与拟进行临床验证的药品同批号。 2.3.6包装、标签和说明书应为国家食品药品监督管理局批准。 2.3.7药学和临床资料是否齐全。 2.3.8GSP证书应注意其批准内容。 2.3.9进口药品检验报告书应注意其结论。 2.3.10管制药品的文件是否齐全。 2.3.11中药保护品种证书所注类别及保护期限。 2.4资料不全的应督促相关单位补齐资料。 2.5形式审查内容 2.5.1根据新药证书、生产批件和试生产批件、批准文号、注册商标批件、包装、标签和说明书实样或报备文件判断是否合法药品,所属药品类别。 2.5.2查询医院信息系统(HIS),判断是否新药。 2.5.3根据药品生产企业许可证和营业执照、GMP证书、药品经营企业许可证和营业执照、GSP证书,判断生产商和销售商是否合法经营。 2.5.4药品的药学、药理、毒理、临床、不良反应等方面的资料是否齐全且有明确结论。
欧洲药品评价局EMEA和欧洲药品质量管理局(EDQM)介绍 一九九四年经欧共体与欧洲议会协商后,以设在法国的欧洲药典委员会秘书处为基础成立了欧洲药品质量管理局(EDQM)。相对于设在英国伦敦主要负责对新药和新生物制品审评的欧洲药品审评委员会(EMEA),EDQM主要功能之一是对上市后的仿制药品的监督管理,其主要监管手段是对产品的Certification of Suitability和对通过欧洲各国家官方药品检验所(OMCL)之间的欧洲网络系统来对药品的市场监督。 EMEA (European Medicines uation Agency)翻译为欧洲药品评价局,其机构正在改革 变化中,首先,EMEA将从现有的“欧洲药品评价局(European Medicines uation Agency,EMEA) 更名为“欧洲药品局(European Medicines Agency,EMA)“。欧洲药品质量理事会EDQM (European Directorate for the Quality of Medicines)作为另一重要欧洲官方药管机构,欧洲 药品质量管理局是由欧洲药典委员会技术秘书处演化而来,它有很多职能,具体职能如下: 1、欧洲药典委员会的技术秘书处提供技术支持 2、负责欧洲药典及相关产品的出版与发行 3、负责化学药物标准品和生物制品标准品的制备与销售 4、负责对欧洲药典各论的适用性认证 5、负责构建欧洲官方药品检验实验室网络,承担生物制品批签发与上市药品的监督任务。 EMEA和EDQM之间的关系? 欧洲药品评价局EMEA(European Agency for the uation of Medicinal Products)是欧洲官方 药管机构之一,它有很多职能,其中很重要的一点就是负责药品(制剂)上市核准程序;而欧洲药品质量理事会EDQM(European Directorate for the Quality of Medicines)作为另一重要欧洲官方药事管理机构,它有很多职能,如:建立药品的质量标准以供欧洲药典委员会使用,制备标准品CRS,执行COS程序最终颁发COS证书等等。二者都是欧洲官方药管机构,它们有着密切的合作伙伴关系。 每个欧盟成员国(总计25个)在EMEA人用药和兽药科学咨询委员会中有一名代表,目 前的委员会为专卖药品委员会和兽药委员会。专卖医药产品委员会(Committee for Proprietary Medicinal Products)将更名为人用药委员会(Committees for Human Medicinal Products,
新药准入审批制度 一、新药审批会议制度 1、药事管理委员会每年度召开1--2次会议,审批新药申请和老药淘汰,参加会议人数须超过应到人数的一半以上。 2、药剂科将初选合格的申请表整理汇总,编制"药事管理委员会讨论药品目录",标明每个品种的商品名、化学名、剂型、规格、报销属性、申请科室、主要用途、生产厂家、参考价格、批准文号、现有同类产品等属性。 3、参会人员每人1份目录,听取申请科室委员对药品有关内容介绍,经提出讨论意见后,在目录上无记名投票。 4、得票超过参会人数半数者为批准购入和淘汰药品,但原则上应控制每次增加和淘汰品种超过30﹪个。 5、药事管理委员会可根据申请情况确定是否请申请人到会答辩。 二、新药申请及审批程序 (一)临床科室申请 1、凡申请购入医院从未使用过,或因各种原因停用半年以上的药品,均应由科室申请,新药还包括不同剂型或不同规格的品种。 2、专科用药须由相应的专科申请,一般情况下西药由西医科室申请,中药由中医科室申请,专科用药由专科科室申请。 3、申请医师须具备主治医师以上职称,申请购入的药品须经全科讨论,科主任签字后附有关资料交药剂科。所附资料包括由生产厂家提供的新药证书、药品说明书、GMP证书、药检报告、药价批单、临床研究报告以及其他用以证明该药优势的文件资料等。
4、申请表中各项内容应填写完整,剂型、类别栏目中应注明是否为何地医保药品及国产、进口、合资等属性。 5、科室主任应对所申请药品用量负责,购入后造成积压浪费由申请科室承担责任,药剂科定期报告用药情况。 (二)医务部审核并签署意见 1、医务部审核申请表及所附资料,内容不全的申请无效。 2、符合下列条件的品种方可提交药事管理委员会审批: (1)本院尚未购入使用的新成分的药品。 (2)注射剂必须是GMP产品。 (3)已有同成分进口产品,因价格因素可申请一种国产药并存(国产药品价格须便宜30%以上)。 (4)现有品种为自费药品时,可申请同种医保用药品种(例如:注射剂自费-口服剂型医保;进口药自费-国产药医保)。 (5)同成分品种,可在质量相同的情况下,以价格低取代价格高的品种。 (6)不同科室申请由不同厂家生产的同一成分药品时,医务部和药剂科共同只认可一份申请。 3、按上述条件初审合格后,医务部主任签署意见。 (三)药剂科审查资料并编制"药事管理委员会讨论药品目录" 1、药剂科和监审科对新药证书、药检报告、药价批单、药品说明书、GMP证书等资料的有效性进行审核,如有疑义可退回申请。 2、按照申请科室汇总,编制"药事管理委员会讨论药品目录",内容包括每个申请品种的商品名、化学名、剂型、规格、报销属性、申请科室、主要用途、生产厂家、参考价格、批准文号、现有同类产品等属性、。
e m e a生物利用度和生物等效性研究指导原则问答 This model paper was revised by the Standardization Office on December 10, 2020
审评四部审评七室陈俊春高晨燕 EMEA自2002年对《生物利用度和生物等效性研究指导原则》(以下简称EMEA指导原则)修订后,于2006年7月发布了《生物利用度和生物等效性研究指导原则问答》(以下简称EMEA指导原则问答)对该原则的一些重要部分作出解释。以下就其问答全文结合我国的《化学人体生物利用度和生物等效性研究技术指导原则》(以下简称我国指导原则)与EMEA指导原则做一简介。 1.、生物等效判定时对Cmax的要求 EMEA指导原则的生物利用度等效评价要求Cmax比值的90%置信区间在–范围内。特殊情况下,如药物治疗窗窄,则可接受的区间范围应更窄。仅在特定情况下,才可接受更宽的区间范围,如-;而且该区间应事先确定,即在试验设计时应考虑到接受大于常规区间范围的情况,事后扩大原方案中确定的可接受区间的做法不可取;并应证明该范围对于病人更换时在安全和有效性方面的合理性。 EMEA指导原则在此提及的增加Cmax比值(非AUC)可接受区间范围的情况并不多见,并且仅扩大了一点,仍窄于我国指导原则中规定的范围-。扩大时仅限于以下情况:1)该药物安全性和有效性的PK/PD相关性资料足以显示Cmax可接受区间的扩大不会显着影响其临床药效。 2)如PK/PD资料不充分,临床安全性和有效性资料可以作为替代,但这些资料仅限于该研究药物。 3)药物在个体内的生物利用度具有高变异性。EMEA和我国的指导原则都对高变异性药物做了定义(即:个体内变异系数大于30%),但是要评价其个体内变异性需要设计重复试验。
X X X酒店L O G 购买药品委托书 A U T H O R I Z A T I O N O F M E D I C I N E P U R C H A S E 本人, , 入住晋江佰翔世纪酒店房间,授权该酒店员工按照本人如下要求为本人购买药品,而该酒店和酒店员工无需承担任何责任。 I, , the guest of Room of Fliport Shiji Hotel Jinjiang, would like to authorize the staff of this hotel to purchase the medicine for me according to my requirement as stated below. The hotel management and the staff will not take any liability whatsoever caused by this. *************************************************************** 1) 药品名称: ?Medicine: 1. 所需药品的说明: ?Description of the medicine expected: *************************************************************** 客人姓名/ Guest name: 房间号码/ Room No.: 证件号码/ PP or ID No. : 经办人/ Handled by: 日期/ Date:
北京医院新药采用审批程序 1.申请: 1)新药申请须副主任医师以上职称者负责填写《北京医院新药采用申请表》,科室的正主任签字同意。专科用药须由相应的专科申请,中成药一般应由中医科提出申请,西医科室申请中成药须由中医专家就方解、功能主治等签署意见。 2)申请表内容包括:药品基本信息、申购理由等。 3)申请表交药学部药品供应室主任。 4)药品供应室主任确认表格填写无误、内容完整后,在表格上标注申请编号和受理日期。 2.形式审查: 1)药品采购员凭《北京医院新药采用申请表》对已受理的申请进行形式审查。 2)药品采购员按《北京医院新药采用申请表》上记录的药品生产商和销售商的联系方式与其联系,索取资料。 (1)一般化学药品应索取下列资料:药品生产企业许可证和营业执照、药品生产批文或试生产批件、新药证书、注册商标批件、物价批文、GMP证书、法定质量标准、药品出厂检验报告书、包装、标签和说明书实样、药品的药学(药学、药理、毒理)资料和临床(临床、不良反应)资料、药品经营企业许可证和营业执照、GSP证书、《药品生产企业质量保证情况调查表》。 (2)进口药品还应索取进口药品注册证、中文说明书、进口药品通关单、进口药品检验报告书。 (3)进口生物制品还应索取生物制品进口批件或生物制品进口批签发证。 (4)麻醉药品、医用毒性药品、精神药品、医用放射性药品应按药学部的有关规章制度索取资料。 (5)中药成药涉及保护品种的还应索取中药保护品种证书。 (6)以上文件可为复印件,所有文件均须加盖该企业的原印章。 3)形式审查应关注以下方面: (1)证书、批件应注意颁发机关、编号和有效期至; (2)国家定价品种应有商务部文件,企业自定价品种应有北京市的物价备案文件; (3)GMP证书应注意其批准内容(全厂认证或生产线认证); (4)质量标准为国家标准或地标升国标; (5)检验报告书是否与拟进行临床验证的药品同批号; (6)包装、标签和说明书应为FSDA批准; (7)药学和临床资料是否齐全; (8)GSP证书应注意其批准内容; (9)进口药品检验报告书应注意其结论; (10)管制药品的文件是否齐全; (11)中药保护品种证书所注类别和保护期限。 4)资料不全的应督促相关单位补齐资料。 5)形式审查的内容包括: (1)根据新药证书、生产批件或试生产批件、批准文号、注册商标批件、包装、标签和说明书实样或报备文件判断是否合法药品,所属药品类别; (2)查询北京医院HIS系统,判断是否新药; (3)根据药品生产企业许可证和营业执照、GMP证书、药品经营企业许可证和营业执照、GSP证书,判断生产商和销售商是否合法经营;
新药注册特殊审批管理规定 第一条为鼓励研究创制新药,有效控制风险,根据《药品注册管理办法》,制定本规定。 第二条根据《药品注册管理办法》第四十五条的规定,国家食品药品监督管理局对符合下列情形的新药注册申请实行特殊审批: (一)未在国上市销售的从植物、动物、矿物等物质中提取的有效成份及其制剂,新发现的药材及其制剂; (二)未在国外获准上市的化学原料药及其制剂、生物制品; (三)治疗艾滋病、恶性肿瘤、罕见病等疾病且具有明显临床治疗优势的新药; (四)治疗尚无有效治疗手段的疾病的新药。 主治病证未在国家批准的中成药【功能主治】中收载的新药,可以视为尚无有效治疗手段的疾病的新药。 属于(一)、(二)项情形的,药品注册申请人(以下简称申请人)可以在提交新药临床试验申请时提出特殊审批的申请。 属于(三)、(四)项情形的,申请人在申报生产时方可提出特殊审批的申请。 第三条国家食品药品监督管理局根据申请人的申请,对经审查确定符合本规定第二条情形的注册申请,在注册过程中予以优先办理,并加强与申请人的沟通交流。 第四条申请人申请特殊审批,应填写《新药注册特殊审批申请表》(附件1),并提交相关资料。 《新药注册特殊审批申请表》和相关资料应单独立卷,与《药品注册管理办法》规定的申报资料一并报送药品注册受理部门。 第五条药品注册受理部门受理后,应将特殊审批申请的相关资料随注册申报资料一并送交国家食品药品监督管理局药品审评中心。 第六条国家食品药品监督管理局药品审评中心负责对特殊审批申请组织审查确定,并将审查结果告知申请人,同时在国家食品药品监督管理局药品审评中心上予以公布。 (一)属于本规定第二条(一)、(二)项情形的,国家食品药品监督管理局药品审评中心应在收到特殊审批申请后5日进行审查确定; (二)属于本规定第二条(三)、(四)项情形的,国家食品药品监督管理局药品审评中心应在收到特殊审批申请后20日组织专家会议进行审查确定。 特殊审批申请的审查确定时间包含在《药品注册管理办法》规定的技术审评工作时间。 第七条国家食品药品监督管理局药品审评中心对获准实行特殊审批的注册申请,按照相应的技术审评程序及要求开展工作。负责现场核查、检验的部门对获准实行特殊审批的注册申请予以优先办理。 第八条获准实行特殊审批的注册申请,申请人除可以按照国家食品药品监督管理局药品审评中心的要求补充资料外,还可以对下列情形补充新的技术资料: (一)新发现的重大安全性信息; (二)根据审评会议要求准备的资料; (三)沟通交流所需的资料。
欧洲药物管理EDMF&CTD基本介绍 EDMF文件简介: 欧洲药物管理档案(EDMF,即 European Drug Master File)是药品制剂的制造商为取得上市许可而必须向注册当局提交的关于在制剂产品中所使用的原料药的基本情况的支持性技术文件。它的申请必须与使用该原料药的制剂的上市许可申请同时进行。当原料药物的生产厂家(ASM,即 The Active Substance Manufacturer)不是药品制剂上市许可证的申请人时,也就是说当制剂生产厂家使用其它厂家生产的原料药物生产制剂时,为了保护原料药物的生产及质量管理等方面有价值的技术机密而由原料药物的生产厂家提交给欧洲官方机构的文件。分为公开部分和保密部分。与美国FDA的DMF涵概药品生产的全过程CMC(Chemistry, Manufacturing and Control)不同,欧洲DMF则主要强调第一个C,即Chemistry。具体的说,EDMF 的主要内容是药物及其相关杂质的化学,包括化学结构及结构解析、化学性质、杂质及其限度、杂质检查等等。 EDMF的适用范围: EDMF适用于以下三类原料药的申请: --仍由专利保护的新的原料药,并且这种原料药没有包括在欧洲药典或任何一个成员国的药典之中; --已过专利保护期的原料药,并且这种原料药没有包括在欧洲药典或任何
一个成员国的药典之中; --包括在欧洲药典或任何一个成员国的药典之中的原料药,当这种原料药使用一个可能留下药典专论没有提到的杂质并且药典专论不能足够控制其质量的方法生产时。EDMF的变动和更新 如果EDMF持有人需要对EDMF的公开部分和保密部分做出变动,则任何变动均要向主管当局或EMEA上报,并通知所有申请人。若仅是修改EDMF 的保密部分,并且生产采用的质量标准和杂质范围均没有发生改变,修改信息只需提供给主管当局;如果需要修改EDMF的公开部分,此信息必须提供给其他申请人和使用此EDMF的药品上市许可证的持有人,所有涉及到的申请人将通过适当的变更程序修改他们的上市许可证申请文档。 EDMF持有人应对EDMF文件在现行的生产工艺,质量控制,技术发展法规和科研要求方面保持内容更新。如果没有任何改变,在欧盟内使用此EDMF的第一个五年后,EDMF持有人应正式声明EDMF文件的内容仍然是不变和适用的,并提交一份更新的申请人或制剂生产厂家的名单。 EDMF的递交程序: 根据欧洲药物管理档案程序的要求,EDMF只能在递交制剂药品上市许可证申请时递交,并且只有欧洲的制剂生产厂家及其授权的代表(如,进口商)才能递交EDMF。
购买药品的类别和药品标识详解 《国家基本医疗保险和工伤保险药品目录》(简称《药品目录》) 基本医疗保险药品(西药和中成药)分甲、乙类。 工伤保险药品不分甲、乙类。 处方药是必须凭执业医师或执业助理医师处方才可调配、购买和使用的药品。 非处方药是不需要凭医师处方即可自行判断、购买和使用的药品。处方药和非处方药是管理上的界定。 非处方药主要用于治疗各种消费者容易自我诊断、自我治疗的常见轻微疾病。 根据药品的安全性,非处方药分为甲、乙两类。 应在药师指导下购买使用甲类非处方药,可自行选择购买和使用乙类非处方药。 上市的中西药品中哪些作为处方,哪些作为非处方,由国家药品监督管理部门组织有关部门和专家进行审核批准的。 药品需经相应药品监督管理部门(食品药品监督管理局-国务院的直属部门)批准方可销售。 外用药品红色甲类非处方药白色乙类非处方药
红色甲类非处方药绿色乙类非处方药 产品包装上的(R)和(TM)标志代表什么意思? TM是TRADEMARK的缩写,美国的商标通常加注TM,并不一定是指已注册商标。而R是REGISTER的缩写,用在商标上是指注册商标的意思. 注册标记包括(注外加○)和(R外加○)。使用注册标记,应当标注在商标的右上角或者右下角。因此,TM与R是不同国家的商标标记,没有特别的关系. 几种药品标识的含义 1、药品通用名称(药品通用名):列入国家药品标准的药品名称。 2、药品商品名称(药品商品名):经国家(食品)药品监督管理
局批准,在《药品注册证》“商品名称”栏中列示的名称。 3、药品注册商标:经国家工商行政管理总局商标局批准注册,受《商标法》保护的药品标识。 4、药品未注册商标:企业未经注册、自行使用,国家不禁止(《商标法》第四十八条规定的除外,以下同),也不受法律保护的药品标识。 易引起混淆的两个概念 如上所述,药品商品名称(药品商品名),是经国家(食品)药品监督管理局批准,在《药品注册证》“商品名称”栏中列示的名称。药品未注册商标,是企业未经注册、自行使用,国家不禁止,也不受法律保护的药品标识,在药品的包装、标签和说明书上通常在其标识的右上角用“TM”标示,如“××××丸?”。 常见有人把“药品未注册商标”称为“药品商品名”,有的涉药企业在药品标价签或发票和有关单据上把“药品未注册商标”作为“药品商品名”标示,严格地说,都是混淆了这两个名词的概念。 只有经国家(食品)药品管理局批准,在《药品注册证》“商品名称”栏中列示的名称才能叫做“商品名”,其他的所有名称均不能叫做“商品名”。 药品包装、标签和说明书管理上的盲点 根据《药品包装、标签和说明书管理规定》第六条规定,“药品
新药申报流程 (一)、报临床 1号资料(药品名称):表述药品名称,阐明名称命名理由或来源,新剂型、新命名、应附上药典委员会命名的复函。 2号资料(证明性文件): 1、申报人的资质:生产厂的“三证”(注意名称地址的一致和有效期);除生产企业外的合作研究方同时申请新药证书需提供有效的营业执照、研究单位的法人证书及其变更登记证明 2、专利查询报告、不侵权保证书(加盖所有申请人的红章) 3、属特殊药品的须有SFDA安监司的立项批件 4、制剂用原料药的合法来源(上省批件或进口注册证、原料厂家的三证、供货协议、购买发票、质量标准、出厂报告书及自检报告书),向经销单位购买的,还需提供经销商与原料厂的供货协议 5、申请人同时申报原料与制剂的则不提供“4”的资料,若同时申请原料和制剂的厂家不是同一家,则须提供原料厂与制剂厂之间的供货协议或合作开发协议以及原料药受理单。(相同制剂,原料药只能和一个申请人合作申报)。 6、拟申请商品名的提供商标查询单或商标注册证 7、直接接触药品的包装材料或容器的供应商资质、包材注册证、报告书、质量标准 8、委托试验:应提供委托合同,并附该机构合法登记证明、必要的
资质证明。 3号资料(立题目的与依据):参考药审中心的指导原则要求 4号资料(对主要研究结果的总结及评价):参考药审中心的指导原则要求 1 / 11 5号资料(药品说明书、起草说明及相关参考文献):格式符合24号令要求,附参考的文献资料6号资料(包装、标签设计样稿):报临床不提供 7号资料(药学研究资料综述):参考相关的技术指导原则 8号资料(原料药生产工艺的研究资料及文献资料;制剂处方及工艺的研究资料及文献资料):参考相关的技术指导原则 9号资料(确证化学结构或者组份的试验资料及文献资料):参考相关的技术指导原则(报告书及图谱上盖章) 10号资料(质量研究工作的试验资料及文献资料):参考相关的技术指导原则 11号资料(药品标准及起草说明):起草说明所依据的试验数据至少应加上中试三批样品的结果及临床用样品的结果,或者应加上做验证的多批样品的数据。 12号资料(样品的检验报告书):申报样品的自检报告书(1~3批),13号资料(原料药、辅料的来源及质量标准、检验报告书): 原料合法来源证明文件一套(同2号资料中的原料资质)辅料的来源
按照《新药审批办法》第五章的规定:“新药的申报与审批分为临床研究和生产上市两个阶段。初审由省级药品监督管理部门负责,县市由国家药品监督管理局负责。” 现结合图1新药申报与审批基本程序示意图介绍该程 1.申报单位填写新药临床研究(或生产)申请表,连同申报的技术资料和样品报省、自治区、直辖市药品监督管理部门。省级药品监督管理部门进行初审,即对新药的各项原始资料是否齐全进行审查;同时,派员对试制条件进行实地考察,填写考察报告表。对已报齐所有应报资料的,正式通知申报单位收审;同时将样品和技术资料转省、自治区、直辖市药品检验所审核。对应报资料不全的,予以退审,将申请表和资料退回申报单位并提出退审理由。 2.省、自治区、直辖市药品检验所按新药审批各项技术要求完成对申报资料的审查和样品的检验。 药检所的审核系指对新药的药学(包括药理、毒理)研究资料进行审查和对样品进行实验检验;不包括为申报单位进行新的检测方法的研究。药检所审核完毕后,提出质量标准和对药学(包括药理、毒理)方面的综合审查意见,送省级药品监督管理部门。 3.省级药品监督管理部门初审通过同意上报的,在新药临床研究(或生产)申请表签署意见,连同申报的技术资料一式5份报国家药品监督管理局注册司进行形式审查。 3’.新生物制品和按《新药审批办法》第二十六条所列新药,由申报单位填写申请表,连同申报的技术资料一式五份直接报国家药品监督管理局注册司。样品检验和质量标准复核由中国药品生物制品检定所负责。4.国家药品监督管理局注册司经形式审查合格的,向申报单位发出收取审评费的通知。同时交药品审评中心安排技术审查、审评委员会审评及必要的复核等工作。 4.形式审查不合格的,予以退审。 5.技术审评通过后,将建议批准的或退审的审评报告及意见,报国家药品监督管理局药品注册司。