
分子的几何构型优化计算(2)
m ents (2)
a n98)
1.优化目的:
对分子性质的研究是从优化而不是单点能计算开始。这是因为我们认为在自然情况下分子主要以能量最低的形式存在。只有能量最低的构型才能具有代表性,其性质才能代表所研究体系的性质。在建模过程中,我们无法保证所建立的模型有最低的能量,所以所有研究工作的起点都是构型优化,要将所建立的模型优化到一个能量的极小点上。只有找到合理的能够代表所研究体系的构型,才能保证其后所得到的研究结果有意义。
分子性质研究的一般模式:
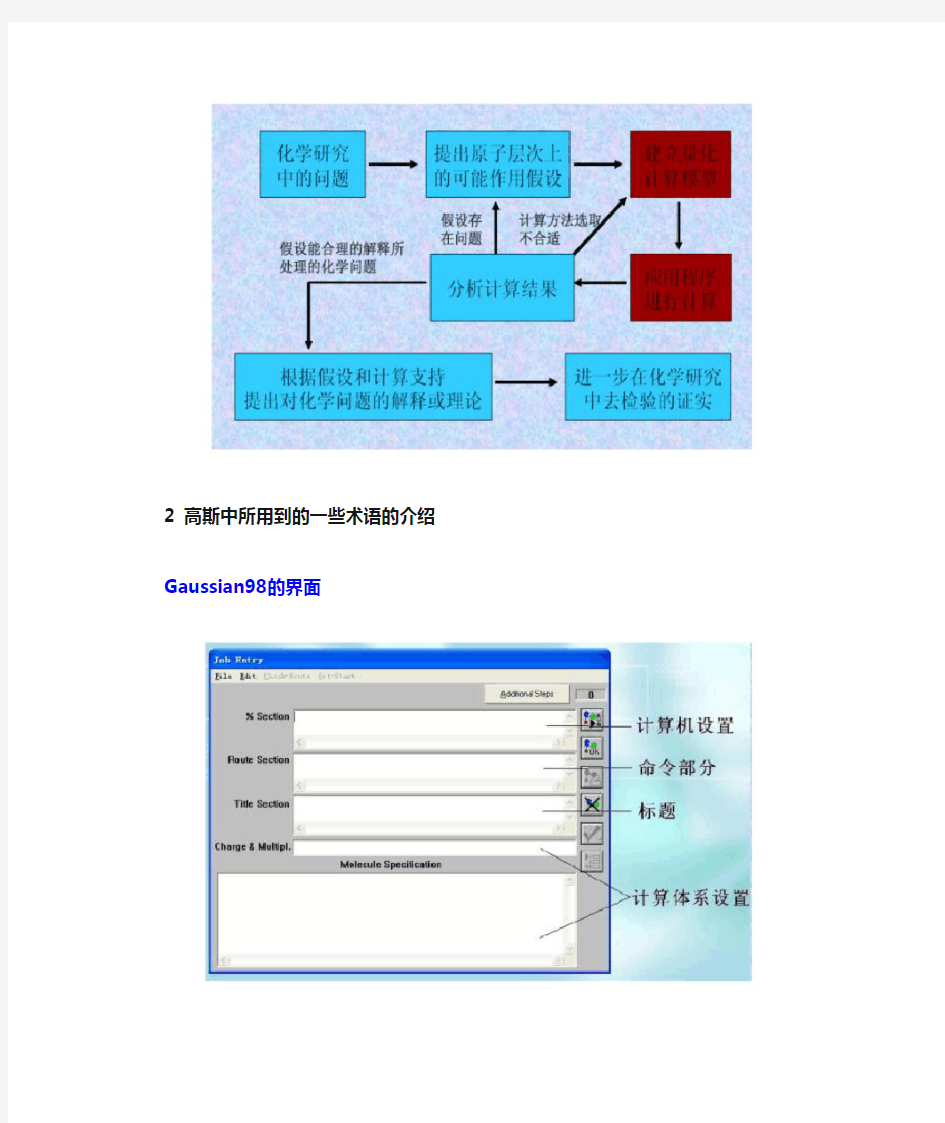
2 高斯中所用到的一些术语的介绍
a n98的界面
2.1势能面
在不分解的前提下,分子可以有很多个可能的构型,每个构型都有一个能量值,所有这些可能的结构所对应的能量值的图形表示就是一个势能面,势能面描述的是分子结构和其能量之间的关系,以能量和坐标作图。根据分子中的原子数和相互作用形式,有可能是二维的,也有可能是多维的。势能面上的每一个点对应一个具有一个能量的结构。能量最低的点叫全局最小点,局域最小点是在势能面上某一区域内能量最小的点,一般对应着可能存在的异构体。鞍点是势能面上在一个方向有极大值而在其他方向上有极小值的点,通常对应的都是过渡态。优化的目的就是找到势能面上的最小点,因为这个点所对应的构型能量最低,是最稳定的。
2.2确定能量最小值
构型优化就是找体系的最小点或鞍点。能量的一阶导(也就是梯度,注意在数学中,一阶导表示着函数的变化趋势,一阶导为零就表明找到了极值点,这是确定最小值的数学基础)是零,这表明在这个点上的力也是零(因为梯度的负值是力)。我们把势能面上这样的点称为静态点(也就是上面所说的极小点)。所有成功的优化都会找到一个静态点,虽然有时找到的静态点并不是想要的静态点。
程序从输入的分子构型开始沿势能面进行优化计算,其目的是要找到一个梯度为零的点。计算过程中,程序根据上一个点的能量和梯度来确定下一步计算的方向和步幅。梯度其实就是我们所说的斜率,表示从当前点开始能量下降最快的方向。以这种方式,程序
始终沿能量下降最快的方向进行计算,只至找到梯度为零的点。而梯度为零表明能量已是最小,所以这个点就是我们所要找的具有最小能量的结构。很多程序还可以计算能量的二阶导,所以很多和能量的二阶导相关的性质(如频率计算)也可以得到。
2.3 计算收敛的标准
优化计算不能无限制的进行下去,判定是否可以结束优化计算的判据就是我们现在所要了解的收敛标准。需要特别强调的是这个标准规定的是两个S CF计算结果间的差距,当计算出的这两个能量值的差落在程序默认的标准之内时,程序就认为收敛达到,优化结束。而单点能计算中也有一个收敛标准,这个收敛标准是用于判定C F计算是否完成。SCF计算是一个迭代过程,假定一个解,带入到方程中,求出一个解,再将这个解带入到方程中,如此循环,直到两次解的差落在程序默认的范围之内,SCF计算完成。在这里强调这二者区别的原因是:优化是高斯计算中最易出错的地方,有时OPT=TIGHT可以帮助我们解决这个问题,所以我们要注意这个命令和S CF=TIGHE的区别。
四个收敛标准:(真正的0难以达到,程序给出了 个判断标准)
在优化过程中,有时会出现只有前两项收敛(YES表示已收敛,NO表示不收敛),这种结果是可以接受的。这是因为高斯程序默认:当计算所得的力已比收敛指标小两个数量极时,即使n t值仍大于收敛指标,也认为整个计算已收敛。这种情况对大分子(具有较平缓的势能面)比较常见。
3. 基本输入格式和输出解释
注意这是程序默认要计算的次数,优化计算有可能提前完成也有可能在默认的次数内不能完成计算。如是后者,通常是用i ew打开输出文件,这时所得的结构对应着输出文件中第 7次计算的结果,在这个结构的基础上再接着进行优化计算;1表示这是第一次优化计算)
Old X 结构旧的变量值, New X 优化计算对于该位置要达到的新的变量值。
四个收敛标准,NO 表示还未收敛。YES表示已收敛。在这一项输出之后给出计算所得的分子结构参数。然后是一个单点能计算,该计算会给该结构的能量(以e e为单位)。收敛后会有以下的输出。
以下输出是以内坐标形式给出优化好的结构。从中可得到需要的参数(键长,键角和二面角)
剩下的输出的部分是优化结构的布局分析(用POP=FULL命令会有详细的输出),分子轨道,原子电荷和偶级距。
4高斯中自带的练习(通常都在a mple文件夹和r cise文件夹中)s e3.1异构体优化练习
s e3.2异构体优化练习考察结构和能量的关系
s e3.3优化练习,考察取代基对分子键长,电荷等的影响
s e3.4优化练习在这个练习中用了C C这个命令助其收敛。其意思是:SCF 计算一开始就使用正常级别的积分精度。通常情况下是先用中等积分精度粗算,稍后再转换为正常积分精度计算。
s e3.5优化核磁计算练习。先优化,算核磁;优化TMS,算核磁。两个值相减后就可以得到和实验值相比较的结果。计算时通常都是先优化,然后在更高的计算方法上算性质(如核磁)。但要注意:频率计算的优化和频率计算必须在同一方法上进行,否则计算结果无意义。本练习是L YP/6-31G(D)的基础上,用HF/6-311+G(2D,P)方法算核磁。
Exe s e3.6 C60优化。记得在做练习时,报错,如是,在命令行中加N OSYM即可。因为体系默认在优化时保持对称性。由于所算体系对称性非常高,优化过程中结构稍有变化,对称性就被破坏。程序就认为不满足限制条件,计算终止。加后,去掉对称性限制,允许结构的变化,所以可计算。
s e3.7过渡态优化计算。这个计算会报错。这个命令要求在每一个输入的结构后都有一个指定特定输出的内容[也就是输入内容中的4.5]
Ex s e3.8比较矩阵坐标,直角坐标和内坐标的优化优势。现在普遍认为内坐标在优化方面比矩阵坐标,直角坐标有优势。所以在默认情况下程序会将输入的坐标自动转化为内坐标形式进行优化。如想用矩阵坐标或直角坐标优化,可用OPT命令实现,参见OPT关键词说明。
5 当优化遇阻时常用的解决办法
1) 看所给的初始构型是否合理,这是初学者最易犯的错误。解决办法:检查初始构型的空间构型,然后先用半经验方法或小基组(如STO-3G)优化,然后再用大基组优化。
2 ) 一般的问题用1的方法就可以解决,如还不行可以用OPT命令增大循环次数,减少步长和提高收敛精度来解决。详见G98或G03手册P T关键词。注意报错其实不是错误,而只是在程序默认的次数内未完成优化任务,用L E命令增大循环次数即可解决这类报错。
3) 对于过渡态优化,由于分子构型需要手动调整,所以更难给出合理的初始构型,通常在命令行里加,该命令意思是在优化前先进行一个频率计算以获得用于指明优化方向的力常数。这个方法也可用于基态难以完成的优化。对于基态的优化,还可用命令来获得力常数,前提是在低一级别的计算水平上作了频率计算,且保留了检查点文件。
L,最无奈的办法,但及耗时,对于我们所要处理的四五十个原子的体系,用单机进行这样的计算太费时间了,已不具有任何实际的意义。这个命令的意思是在每一步优化前都要做一个频率计算获得指明下一步计算的力常数。
5.当计算因外因(如停电等)意外中止时,如果保留有检查点文件可用R ESTA T命令继续这个计算,用c k命令后,分子的电荷,多重度和结构说明部分都不需要,如果是用o m=check命令,则需要有电荷和多重度的说明。详细格式如下:
这是一个关于过渡态优化因停电终止而的例子。 t和
c k 命令必须要连用。rwf 文件是用来指定临时交换文件空间大小的,通常情况下程序默认在R ATCH文件中产生一个约2G的临时文件,对一般计算来说,这个存放临时文件的空间够了。但对于大的频率计算,大的从头算法(如MP2等)和CIS计算等一些会产生大的临时交换数据的算法来说,可能会因F文件空间不够而报错,这时可通过这个命令格式加大临时数据的存放空间,据说对于单机版格式的分区,程序最大可允许设定8个临时文件,总空间是 G-24G(具体视操作系统而定,据说XP系统可达24G,但W98系统只能达到 G);对于NTS格式,可产生的单个文件约为 ,总共可产生 2G的临时文件空间。[注:关于RWF的大多介绍来自大话西游,量子化学研究小组和厦大论坛,自己未试过] Geom=(Check, Step=n)这个命令要求从已停止的优化计算的第步开始重新优化。一般在失败的优化中有一步非常接近收敛值,但在这一步后又偏移了的情况下用这一命令,有时还要加上促使其收敛。
6 实验内容
●练习高斯中自带的几何构型优化例题,熟悉几何构型优化的基本方法
●对乙烯醇异构体(或自己感兴趣的简单分子)进行几何构型优化,将理论计算结果与
文献实验结果分析比较,初步了优化方法的特点。
简化解析几何运算的技巧 中学解析几何是将几何图形置于直角坐标系中,用方程的观点来研究曲线,体现了用代数的方法解决几何问题的优越性,但有时运算量过大,或需繁杂的讨论,这些都会影响解题的速度,甚至会中止解题的过程,达到“望题兴叹”的地步.特别是高考过程中,在规定的时间内,保质保量完成解题的任务,计算能力是一个重要的方面.为此,从以下几个方面探索减轻运算量的方法和技巧,合理简化解题过程,优化思维过程. 导,把定量的分析有机结合起来,则可使解题计算量简化,使解题构筑在较高的水平上. [典例] 如图,F 1,F 2是椭圆C 1:x 24+y 2 =1与双曲线C 2的公共焦点,A , B 分别是 C 1,C 2在第二、四象限的公共点.若四边形AF 1BF 2为矩形,则C 2的离心率是( ) A.2 B.3 C.32 D.6 2 答案:D [方法演示] 解析:由已知,得F 1(-3,0),F 2(3,0),设双曲线C 2的实半轴长为a ,由椭圆及双曲线的定义和已知,可得???? ? |AF 1|+|AF 2|=4,|AF 2|-|AF 1|=2a , |AF 1|2+|AF 2|2=12,解得a 2=2,故a = 2.所以双曲线C 2的离心率e = 32=6 2 . [解题师说] 本题巧妙运用椭圆和双曲线的定义建立|AF 1|,|AF 2|的等量关系,从而快速求出双曲线实半轴长a 的值,进而求出双曲线的离心率,大大降低了运算量. [应用体验] 1.抛物线y 2=4mx (m >0)的焦点为F ,点P 为该抛物线上的动点,若点A (-m,0),则|PF | |P A |的最小值为 ________.
分子的几何构型优化计算(2)Molecular Modelling Experiments (2) (Gaussian98) 1.优化目的: 对分子性质的研究是从优化而不是单点能计算开始。这是因为我们认为在自然情况下分子主要以能量最低的形式存在。只有能量最低的构型才能具有代表性,其性质才能代表所研究体系的性质。在建模过程中,我们无法保证所建立的模型有最低的能量,所以所有研究工作的起点都是构型优化,要将所建立的模型优化到一个能量的极小点上。只有找到合理的能够代表所研究体系的构型,才能保证其后所得到的研究结果有意义。 分子性质研究的一般模式: 2 高斯中所用到的一些术语的介绍 Gaussian98的界面
2.1势能面 在不分解的前提下,分子可以有很多个可能的构型,每个构型都有一个能量值,所有这些可能的结构所对应的能量值的图形表示就是一个势能面,势能面描述的是分子结构和其能量之间的关系,以能量和坐标作图。根据分子中的原子数和相互作用形式,有可能是二维的,也有可能是多维的。势能面上的每一个点对应一个具有一个能量的结构。能量最低的点叫全局最小点,局域最小点是在势能面上某一区域内能量最小的点,一般对应着可能存在的异构体。鞍点是势能面上在一个方向有极大值而在其他方向上有极小值的点,通常对应的都是过渡态。优化的目的就是找到势能面上的最小点,因为这个点所对应的构型能量最低,是最稳定的。 2.2确定能量最小值 构型优化就是找体系的最小点或鞍点。能量的一阶导(也就是梯度,注意在数学中,一阶导表示着函数的变化趋势,一阶导为零就表明找到了极值点,这是确定最小值的数学基础)是零,这表明在这个点上的力也是零(因为梯度的负值是力)。我们把势能面上这样的点称为静态点(也就是上面所说的极小点)。所有成功的优化都会找到一个静态点,虽然有时找到的静态点并不是想要的静态点。 程序从输入的分子构型开始沿势能面进行优化计算,其目的是要找到一个梯度为零的点。计算过程中,程序根据上一个点的能量和梯度来确定下一步计算的方向和步幅。梯度其实就是我们所说的斜率,表示从当前点开始能量下降最快的方向。以这种方式,程序
二、判断分子构型——价层电子对互斥理论(VSEPR) 现代化学的重要基础之一是分子(包括带电荷的离子)的立体结构。实验测出,SO3分子是呈平面结构的,O—S—O的夹角等于120o,而SO32-离子却是呈三角锥体,硫是锥顶,三个氧原子是三个锥角,象一架撑开的照相用的三角架。又例如SO2的三个原子不在一条直线上,而CO2却是直线分子等等。价层电子对互斥理论用以预测简单分子或离子的立体结构,我们不难学会用这种理论来预测和理解分子或离子的立体结构,并用来进一步确定分子或离子的结构。 价层电子对互斥理论认为,在一个共价分子中,中心原子周围电子对排布的几何构型主要决定于中心原子的价电子层中电子对的数目。所谓价层电子对包括成键的σ电子对和孤电子对。价层电子对各自占据的位置倾向于彼此分离得尽可能地远些,这样电子对彼此之间的排斥力最小,整个分子最为稳定。这样也就决定了分子的空间结构。也正因此,我们才可以用价层电子对很方便地判断分子的空间结构。例如:甲烷分子(CH4),中心原子为碳原子,碳有4个价电子,4个氢原子各有一个电子,这样在中心原子周围有8个电子,4个电子对,所以这4个电子对互相排斥,为了使排斥力最小,分子最稳定,它们只能按正四面体的方式排布。这样就决定了CH4的正四面体结构。 利用VSEPR推断分子或离子的空间构型的具体步骤如下: ①确定中心原子A价层电子对数目。中心原子A的价电子数与配位体X提供共用的电子数之和的一半,就是中心原子A价层电子对的数目。例如BF3分子,B原子有3个价电子,三个F原子各提供一个电子,共6个电子,所以B 原子价层电子对数为3。计算时注意:(ⅰ)氧族元素(ⅥA族)原子作为配位原子时,可认为不提供电子(如氧原子有6个价电子,作为配位原子时,可认为它从中心原子接受一对电子达到8电子结构),但作为中心原子时,认为它提供所有的6个价电子。(ⅱ)如果讨论的是离子,则应加上或减去与离子电荷相应的电子数。如PO43-离子中P原子的价层电子数应加上3,而NH4+离子中N原子的价层电子数则应减去1。(ⅲ)如果价层电子数出现奇数电子,可把这个单电子当作电子对看待。如NO2分子中N原子有5个价电子,O原子不提供电子。因此中心原子N价层电子总数为5,当作3对电子看待。 ②确定价层电子对的空间构型。由于价层电子对之间的相互排斥作用,它们趋向于尽可能的相互远离。于是价层电子对的空间构型与价层电子对数目的关系如下表所示:
分子几何构型优化的初步比较 摘要对《CRC物理与化学手册》[1]第78版中收集的已知实验构型的多原子分子NH3, 在HF、MP2的级别上进行了构型优化的初步比较.优化采用基组STO-3G、6-31G(d、p) 6-311G(d、p), 以及6-311G(2d、p). 对同一方法比较了基组函数, 对同一基组函数则比较了HF 和MP2 方法.结果表明,键长的平均绝对偏差(单位:pm)由大到小的顺序为: MP2/STO-3G, HF/STO-3G, HF/6-311G(d、p),HF/6-31G(d、p),HF/6-311G(2d、p), MP2/ 6-31G(d、p) ,MP2/6-311G(d、p). 引言量子化学优化分子几何构型的理论和应用日趋成熟和程序化[2-4],使研究新型配 合物的物质结构,配合物中间体,反应机理研究的手段向前迈进了大大的一步.可供选择的分子几何构型优化的方法很多,但何种理论方法较为精确,一直是相关研究工作者关心和期望得到明确答案的问题.在一些很高级别的能量计算和发展精确模型化学计算方法中[ 5],这一问题更加突出.可以认为分子几何构型优化的可靠性是一切精确计算的基本保证.由于理论上对于分子几何构型优化尚无规律和系统方法克可循目前只能通过一些系统性的比较来探讨这个问题.对于一些有机小分子构型优化的比较,已有文献报道,但仍不够充分,而对于化学建类型变化较多的无机分子的系统比较则较少.[5 ,6]. 构型优化的计算量往往较大,因此寻找和事先确定适当的构型优化方法,已成为计算化学领域中一个共同感兴趣的问题.本文就是从此问题出发, 以NH3分子构型的优化为基点, 以STO-3G、6-31G(d、p), 6-311G(d、p), 以及6-311G(2d、p)为基组,在HF 和MP2级别上进行全面的理论计算和比较.由于计算数据较多,会引起篇幅较大,本文将不一一列出数据,而是直接报导计算和分析统计结果. 1. 实验数据 不同的实验方法得到的数据有所不同,且实验中有平衡构型和各种振动态(多为基态及低激发振动态)的平动构型r0以及同位素平均构型r e等,鉴于低振动态势阱的非谐性对结构参数的影响一般不大,本文将不区分r e和r0等,而全部按文献[1]数据实录. NH3 为C3v对称点群,:,为了计算输入的方便, 将NH3分子构型中的一些实验参数作了等价转化. 2. 计算方法 分子几何构型优化理论方法建立在对分子势能面认识的基础上,其物理模型和数学模型是十分清晰的.这里不再赘述.本文中,我们对NH3分子分成不同基组和不同方法分别讨论.为了突出氢原子基组中加入p极化函数的必要性,重点比较了不同基组态STO-3G和6-31G(d,p), 6-311G(d、p)等.同时对目前普遍采用的HF 方法和MP2 方法进行了同一基组态计算精度的比较分析.,比较其计算度和精度, 分析其偏差原因. .所有计算用Gaussian-98W[2 ] 成. NH3分子的实验键长多数精确到0.01pm,计算结果也精确至0.01pm. 3. 结果与分析
博文教育讲义 课题:简化解析几何运算方法 教学目标:提高学生简化运算的意识,注意探索简捷运算的技巧,并适时进行有关的规律总结 教学重点:简化运算方法归纳 教学难点:有关的规律总结与运用 教学过程: 解析几何的本质特征是几何问题代数化,就是将抽象的几何问题转化为易于计算的代数问题,这提供了许多便利;但也不可避免地造成许多计算的繁琐,同时对运算能力提出较高要求。其实,只要有简化运算的意识,注意探索简捷运算的技巧,并适时进行有关的规律总结,许多较为繁琐的计算过程是可以简化甚至避免的。 1.回归定义 圆锥曲线的定义是圆锥曲线的本质属性。许多美妙而有趣的性质和结论都是在其定义的基础上展开的,在分析求解时若考虑回归定义,可以使许多问题化繁为简。 例1 过椭圆左焦点倾斜角为 60的直线交椭圆于点B A ,且FB FA 2=,则此椭圆离心率为._____ 解析 本题的常规解法是:联立?? ?? ?+==+)(3,122 22c x y b y a x 再结合条件FB FA 2=求解,运算量大,作为填空题,不划算!如图1,考虑使用椭圆的定义和有关平面几何性质来求解: )2(31)(31B B A A B B A A B B FM '+'='-'+'= )2(31e BF e AF +=, 另一方面,在F C B Rt '?中C F BF C BF '=?='∠260 , 故.2 BF e BF M C C F FM += '+'=于是 =+)2(31e BF e AF 2 BF e BF FM +=, 又FB FA 2=,所以可得.3 2 =e 练习:设12F F ,是双曲线()22 2210,0x y a b a b -=>>的左,右两个焦点,若双曲线右支上存在一点P ,使 () 220.OP OF F P +?= (O 为坐标原点),且123PF PF = ,则双曲线的离心率是( ) 32 31. .32. .312 2 A B C D ++++ 【分析】根据向量加法的平行四边形法则,2=,OP OF OQ + 2OQ F P ∴⊥ 2OQ F P 且必过的中点.可知12PF F ?为直角三角形. 这就为用定义法求离心率创造了条件. 【解析】不妨设双曲线的半焦距c=1,.令 ( ) 21=,3,231PF r PF r a r =∴= - 则,1290,F PF ∠=?但是 O A B A ' B 'F x y M 1 图C C 'x y P O F 1 F 2 Q M
Gaussian程序使用:分子平衡几何构型优化及分子性质计算【实验目的与要求】 计算化学,其本质是对分子体系薛定谔方程所代表的化学理论通过大型计算机程序的求解,模拟化学的各种实验研究。作为一门计算化学实验课程,主要目的是从实际操作出发,掌握程序的使用,以便得到预期的结果。对于所涉及的理论和方法,只要求结合程序的演算能够定性予以理解。本实验主要涉及优化分子几何构型的程序输入及计算结果的解读。 【实验原理】 1. 在结构化学中,曾用“变数分离”方法对于单电子体系(氢原子和类氢离子)的Schr?dinger方程进行精确求解。但是对于多电子的分子体系,由于第i 个电子与其余电子间的排斥能取决于所有电子的坐标,使这种分离变为不可能。但可以在定核近似下将核的运动分离出去后,在固定的核势场中近似求解多电子体系的能量本征方程。 具体做法是,对第i个电子,可以假定一个单电子的分子轨道(单电子近似),并将它用现成的原子轨道线性展开(LCAO近似)。这时,Schr?dinger方程由微分方程变成一个齐次线性的代数方程组。求解该方程组,即求各分子轨道能级及相应的分子轨道展开系数。具体过程是在给定的核坐标下,先猜测一组展开系数(极端情况均为0),代入方程组得到一组新的系数,再代入方程组求解,周而复始,直到前后两组系数相同,称为“自恰场迭代”。这就是HF自恰场分子轨道方法。 2. 量子化学中的基组是用于描述体系波函数的若干具有一定性质的函数。基组是量子化学从头计算的基础,在量子化学中有着非常重要的意义。在量子化学计算中,根据体系的不同,需要选择不同的基组,构成基组的函数越多,基组便越大,计算的精度也越高,计算量也随之增大。 要提高量子化学计算精度,必须加大基组的规模,即增加基组中基函数的数量,增大基组规模的一个方法是劈裂原子轨道,也就是使用多个基函数来表示一个原子轨道。 劈裂价键基组就是应用上述方法构造的较大型基组,所谓劈裂价键就是将价层电子的原子轨道用两个或以上基函数来表示。常见的劈裂价键基组有3-21G、4-21G、4-31G、6-31G、6-311G等,在这些表示中前一个数字用来表示构成内层电子原子轨道的高斯型函数数目,“-”以后的数字表示构成价层电子原子轨道的高斯型函数数目。如6-31G所代表的基组,每个内层电子轨道是由6个高斯型函数线性组合而成,每个价层电子轨道则会被劈裂成两个基函数,分别由3个和1个高斯型函数线性组合而成。 劈裂价键基组能够比STO-NG基组更好地描述体系波函数,同时计算量也比最小基组有显著的上升需要根据研究的体系不同而选择相应的基组进行计算。 【实验内容】 1. 输入的基本组成部分。如图所示:
高中数学解析几何解题方法总结 老师在讲题的时候,经常如未卜先知一般,就知道已知条件里经常存在着一个自己完全不知道的信息;或者分析着分析着,就突然来句:“这道题可以用反证法/数学归纳法……”解法是很精妙,但换你来做,你就是没有意识到要采用这样的方法。我也曾经问过老师,为什么你们当时会想到用这种方法?得到的也往往是“不知道”、“题目做多了就明白了”。 高中数学解析几何解题方法我们先来分析一下解析几何高考的命题趋势: (1)题型稳定:近几年来高考解析几何试题一直稳定在三(或二)个选择题,一个填空题,一个解答题上,占总分值的20%左右。 (2)整体平衡,重点突出:其中对直线、圆、圆锥曲线知识的考查几乎没有遗漏,通过对知识的重新组合,考查时既留意全面,更留意突出重点,对支撑数学科知识体系的主干知识,考查时保证较高的比例并保持必要深度。近几年新教材高考对解析几何内容的考查主要集中在如下几个类型: ① 求曲线方程(类型确定、类型未定); ②直线与圆锥曲线的交点题目(含切线题目);
③与曲线有关的最(极)值题目; ④与曲线有关的几何证实(对称性或求对称曲线、平行、垂直); ⑤探求曲线方程中几何量及参数间的数目特征; 高中数学解析几何解题方法: (3)能力立意,渗透数学思想:一些虽是常见的基本题型,但假如借助于数形结合的思想,就能快速正确的得到答案。 (4)题型新奇,位置不定:近几年解析几何试题的难度有所下降,选择题、填空题均属易中等题,且解答题未必处于压轴题的位置,计算量减少,思考量增大。加大与相关知识的联系(如向量、函数、方程、不等式等),凸现教材中研究性学习的能力要求。加大探索性题型的分量。 在近年高考中,对直线与圆内容的考查主要分两部分: (1)以选择题题型考查本章的基本概念和性质,此类题一般难度不大,但每年必考,考查内容主要有以下几类: ①与本章概念(倾斜角、斜率、夹角、间隔、平行与垂直、线性规划等)有关的题目; ②对痴光目(包括关于点对称,关于直线对称)要熟记解法; ③与圆的位置有关的题目,其常规方法是研究圆心到直线的间隔. 以及其他“标准件”类型的基础题。 (2)以解答题考查直线与圆锥曲线的位置关系,此类题综合性比较强,难度也较大。 预计在今后一、二年内,高考对本章的考查会保持相对稳定,即在题型、题量、难度、重点考查内容等方面不会有太大的变化。
结构的几何构造分析概念 1-1 1、几何组成分析的目的主要是分析、判断一个体系是否几何可变,或者如何保证它成为几何不变体系,只有几何不变体系才可以作为结构。 几何可变体系:不考虑材料应变条件下,体系的位置和形状可以改变的体系。几何不变体系:不考虑材料应变条件下,体系的位置和形状保持不变的体系。 2、自由度:描述几何体系运动时,所需独立坐标的数目。 平面内一个动点A,其位置要由两个坐标 x 和 y 来确定,所以一个点的自由度等于2。平面内一个刚片,其位置要由两个坐标 x 、y 和AB 线的倾角α来确定,所以一个刚片在平面内的自由度等于3。 3、刚片:平面体系作几何组成分析时,不考虑材料应变,所以认为构件没有变形。可以把一根杆、巳知是几何不变的某个部分、地基等看作一个平面刚体,简称刚片。 4、约束:如果体系有了自由度,必须消除,消除的办法是增加约束。约束有三种: 5、多余约束:减少体系独立运动参数的装置称为约束,被约束的物体称为对象。使体系减少一个独立运动参数的装置称为一个约束。例如一根链杆相当于一个约束;一个连接两个刚片的单铰相当于二个约束;一个连接n个刚片的复铰相当于n—1个单铰;一个连接二个刚片的单刚性节点相当于三个约束;一个连接n 个刚片的复刚性节点相当于n—1个单刚性节点。如果在体系中增加一个约束,体系减少一个独立的运动参数,则此约束称为必要约束。如果在体系中增加一个约束,体系的独立运动参数并不减少,则此约束称为多余约束。平面内一个无铰的刚性闭合杆(或称单闭合杆)具有三个多余约束。
6、瞬变体系及常变体系:常变体系概念:体系可发生大量的变形,位移。区别于瞬变体系:瞬变体系概念:体系可发生微小的变形,位移。 7、瞬铰:两刚片间以两链杆相连,其两链杆约束相当(等效)于两链杆交点处一简单铰的约束,这个铰称为瞬铰或虚铰。 2-2平面杆件体系的计算自由度 1、体系是由部件(刚片或结点)加上约束组成的。 2、刚片内部:是否有多余约束。内部有多余约束时应把它变成内部无多余约束的刚片,而它的附加约束则在计算体系的约束总数时应当考虑进去。 3、复铰:连接两个以上刚片的铰结点。连接n个刚片的铰相当于(n-1)个单铰。 4、单链杆:连接两个铰结点的链杆。 5、连接两个以上铰结点的链杆。 连接 n 个铰结点的复链杆相当于(2n-3)个单链杆。 6、平面体系的计算自由度 W :W=3m-(2n+r) m:钢片数 n:单绞数 r:支座链杆数上面的公式是通用的。 W=2J-(b+r) J:结点个数 b:链杆数 r:支座链杆数上面的公式用于完全由铰接的连杆组成的结构体系。 7、自由度与几何体系构造特点: 静定结构的受力分析
Gaussian中分子的几何构型 分子的几何构型 ************************************ 分子的几何构型(Molecular Geometry) ************************************ 分子的平衡构型(molecular equilibrium geometry)是分子电子能量和核间排斥能 量最小时分子的核排列。 分子势能 一个含有N个原子核的非线性分子的几何构型可以用3N-6个独立的核坐标决定,分子 的电子能量,U(q1,q2,…,q3N-6)是这些坐标的函数。 U = Ee +VNN 注意到3个平移和3个转动自由度(线性分子的转动自由度为2)对U是没有贡献的,因 此对一个双原子分子,U的表达式中仅仅保护一个变量,即两个核之间的距离,U?。 对一个多原子分子,U是每两个原子核之间距离的函数,是分子势能面(potential energy surface, PES)的一部分。对某一特定的分子核排列下U的计算被成为单点 (single-point)计算,因为这一计算仅仅涉及到分子PES上的一个点。 一个大分子可能在其PES上有多个极小点,对应于不同的平衡构象和鞍点。 分子构象(molecular conformation)可以通过指定围绕单键的二面角的指得到。在 能量极小点处的分子构象称为构型(conformer)。 几何构型优化 从初始几何构型出发寻找U的极小值的过程称几何构型优化(geometry optimization) 或者能量极小化(energy minimization)。极小化的算法同时计算U和U梯度。 在一个局部最小点,U的3N-6个偏微分都是0。PES上▽U = 0的点称为稳定点(statio nary point)或者判据点(critical point),它可以是极小点,极大点或者鞍点。 除了▽U之外,一些最小化方法使用到U的二阶偏微分,从而生成Hessian矩阵,又称为 力常数(force constant)矩阵,因为d^2U/Qi^2 = fi为力常数。 如果一个稳定点是电子能量面上的一个极小点,其力常数矩阵的所有特征值都是正值 。然而,若一个稳定点是过渡态(transition state, TS),其中一个特征值是负值。 Newton-Rapson Newton-Rapson方法是一种非常有效的寻找多变量函数的局部极小点的算法,它将函 数用Taylor展开到二次项,包括函数的一次和二次微分,并以此作为函数的近似。 Quasi-Newton-Rapson 计算自洽场(self consistent field, SCF)能量的二阶微分是非常耗时的,因此在 优化时经常使用一种修正的方法,即quasi-Newton(或quasi-Newton-Rapson)方法。 这种方法在每一步优化中通过计算梯度对Hessian值进行初始估算。 优化方法 为了优化几何构型,要先对平衡构型做一个估算,通常使用键长和键角的经验值。此外,我们还要选择
用几何性质优化解析几何计算 教学设计 海口市第一中学数学组 李哲慧 2012年12月
《用几何性质优化解析几何计算》教学设计 引言:我们在解决解析几何问题时,常常会遇到计算,而有些题目繁琐的计算影响了我们学习解析几何的感情。同时我们又发现一些题目涉及到平面图形的几何性质,如果利用这些性质,可以优化解析几何计算,但我们的学生常常忽略这些重要的性质,本节课意在遇到可以用几何性质优化计算的问题时,不要忽略几何性质,步入繁琐的计算,甚至解不出题目。 一、教学任务分析 1.学情分析:学生已学完高中数学的全部内容,初步掌握解析几何的基本概念、基本题型、基本方法,但灵活应用基础知识解决综合题的能力较弱,计算能力有 待提高,优化计算意识不强。 2.高考中的解析几何:解析几何属高考必考内容,考题涉及图形的几何性质及计算,主要考察数形结合思想,方程思想,对应和运动变化思想等数学思想,既要 求学生的理解能力、分析问题的能力,同时对计算能力要求很高。 3.展示“优化”计算:通过一些题目的几何性质,得出对题目优化计算的解法,同时与代 数法对比,展示用几何性质优化解析几何计算,提高学生数形结合的 解题能力,提高运算速度。 4.学生参与体验:整个过程师生互动,学生观察、体验,在题目的变式中提高发散思维 能力,在题目的由浅入深变换中感受举一反三。 二、教学目标 1.知识层面:由中点(分点)、中垂线,联系三角形中位线、平行线分线段成比例、圆的几何性质、圆锥曲线定义等,要求学生熟悉掌握图形的几何性质,并能灵 活应用。 2.技能方面:①通过对比加强用几何性质优化解析几何计算的能力; ②通过题目的层层深入,提高学生举一反三的能力; ③通过改变题目部分条件,培养学生的发散思维能力,进而提高探究能力。 3.情感方面:在师生互动与学生的交流中,探究问题的发现,分享成功解决问题的喜悦,开阔视野,提升思维的品质,感受几何性质对解析几何计算的优化. 三、重点、难点 重点:用几何性质优化计算. 难点:1.将代数语言转化为几何语言; 2.用几何性质得出简洁的结论.
实验9 分子结构模型的构建及优化计算 一、目的要求 1.掌握Gaussian 和GaussView程序的使用。 2.掌握构建分子模型的方法,为目标分子设定计算坐标。 3.能够正确解读计算结果,采集有用的结果数据。 二、实验原理 量子化学是运用量子力学原理研究原子、分子和晶体的电子结构、化学键理论、分子间作用力、化学反应理论、各种光谱、波谱和电子能谱的理论,以及无机、有机化合物、生物大分子和各种功能材料的结构和性能关系的科学。 Gaussian程序是目前最普及的量子化学计算程序,它可以计算得到分子和化学反应的许多性质,如分子的结构和能量、电荷密度分布、热力学性质、光谱性质、过渡态的能量和结构等等。GaussView是一个专门设计的与Gaussian配套使用的软件,其主要用途有两个:构建Gaussian的输入文件;以图的形式显示Gaussian计算的结果。本实验主要是借助于GaussView程序构建Gaussian的输入文件,利用Gaussian程序对分子的稳定结构和性质进行计算和分析。 三、软件与仪器 1.软件:Gaussian03、GaussView计算软件,UltraEdit编辑软件。 2.仪器:计算机1台。 四、实验步骤 1.利用GaussView程序构建Gaussian的输入文件 打开GaussView程序,如图9-1所示,在GaussView中利用建模工具(View→Builder→),如图9-2所示,在程序界面元素周期表的位置处找到所需的元素,单击即可调入该元素与氢元素的化合物。 图9-1 GaussView打开时的界面
图9-2点击Builder及双击图标后出现的元素周期表窗口图若要构建像乙烷这样的链状分子,需要先点击工具栏中的按钮,常见的链状分子就显示在新打开的窗口中,如图9-3所示。 图9-3 常见链状官能团窗口图 若要构建像苯、萘等环状结构的分子结构,需要双击工具栏中的按钮,常见的环状有机分子就显示在新打开的窗口中,如图9-4所示。 进行分子的基本构型搭建后,在进行元素及键型、特殊基团的选择,重现构建分子直至构建为所需分子。选定要编辑的原子后,在对原子之间的键长、键角或者二面角进行选定,输入所需要的键长、键角或二面角值。要求学生练习构建H2O、CH4、乙烯和乙醛等分子的构型。 绘制出分子的结构式后,把图形保存成gjf文件(File→Save,取名为*.gjf,注意文件名和路径都不能包含中文字符)。
判断分子空间几何构型的简单方法 电子对数目成键电子对 数目孤电子对数 目 分子的空间 构型 实例 2 2 0 直线型二氧化碳 3 3 0 三角形三氟化硼 2 1 V型二溴化锌4 4 0 四面体甲烷 3 1 三角锥氨气 2 2 V型水 5 5 0 三角双锥五氯化磷 4 1 变形四面体四氟化硫 3 2 T型三氟化溴 2 3 直线型二氟化氙6 6 0 八面体六氟化硫 5 1 四角锥五氟化碘 4 2 正方形四氟化氙以下用G表示电子对数目,V表示分子中所有原子最外层电子数的和,n表示配位原子中除了氢原子以外的其它原子的个数,m表示孤电子对数目,r表示配
位原子中氢原子的个数。 当配位原子中没有氢原子且V≥16时:V=8n+2m,G=m+n 例:CO2分子构型的判断 V=4+6×2=8n+2m,这里n=2,∴m=0, ∴G=m+n=0+2=2,所以CO2的分子构型为直线型 BF3分子构型的判断 V=3+7×3=8n+2m,这里n=3,∴m=0, ∴G=m+n=0+3=3,所以BF3的分子构型为三角形 PCl5分子构型的判断 V=5+7×5=8n+2m,这里n=5,∴m=0, ∴G=m+n=0+5=5,所以PCl5的分子构型为三角双锥 SF4分子构型的判断 V=6+7×4=8n+2m,这里n=4,∴m=1, ∴G=m+n=1+4=5,所以SF4的分子构型为变形四面体 BrF3分子构型的判断 V=7+7×3=8n+2m,这里n=3,∴m=2, ∴G=m+n=2+3=5,所以BrF3的分子构型为T型 SF6分子构型的判断 V=6+7×6=8n+2m,这里n=6,∴m=0, ∴G=m+n=0+6=6,所以SF6的分子构型为八面体 XeF4分子构型的判断
四、化学键理论与分子几何构型 1. (1) ,(I)的稳定性大于(Ⅱ)。 (2) C O O O N O C O O O N O O (I) O C O O N O O C O O O N O O (II) O N O O C O O O N O O C O O (III) O N O O C O N O O C O O (IV) 第(III)式最稳定。 (3) Cu + + NO 2–+ 2H + Cu 2+ + NO + H 2O (4) 若压强降到原来的2/3,则说明3 mol NO 变成2 mol 气态物质: 3NO NO 2 + N 2O ,又由于2NO 2N 2O 4,所以最后的气体总压还要略小于原压的2/3。 2. N N N N N N N N N (IV) (V) (II)、(V)不稳定,舍去,(I)比(III)、(IV)稳定。 N (a)N (b)N (c) N (d)N (e) N (a)—N (b)的键级为5/2~3, N (b)—N (c)的键级为1~3/2, N (c)—N (d)的键级为1~3/2,N (d)—N (e)的键级为5/2~3。 N 5+有极强的氧化性。应在液态HF 中制备N 5+。 3. ArCl + OF + NO + PS + SCl + 键级: 1 2 3 3 2 ArCl +键级最小,最不稳定;虽然NO +与PS +的键级都是3,但NO +是2p —2p 轨道重叠的π键,而PS +是3p —3p 轨道重叠的π键。前者重叠程度大,E π大,所以NO +比PS +稳定,即NO +离子最稳定。 4. (1) B 3N 3H 6 N H H H H H N B N B B H H H H H H N B B H N B N O N O O O N O O (I)(II) N N N N N N N N N N (I) (II) N N N N N (III)
几何结构之折叠、旋转(讲义) ?知识点睛 1.折叠(轴对称)的思考层次 (1)全等变换:对应边相等、对应角相等. (2)对应点与对称轴:对称轴所在直线是对应点连线的垂直平分线.(对应点所连线段被对称轴垂直平分,对称轴上的点到对应点的距离相等) (3)常见组合搭配 ①矩形背景下的折叠常出现等腰三角形; ②两次折叠往往会出现特殊角:45°,60°,90°等. (4)应用,作图(构造) 核心是确定对称轴和对应点,一般先确定对应点和对称轴,然后再补全图形. 特征举例: ①折痕运动但过定点,则折叠后的对应点在圆上; ②对应点确定,折痕为对应点连线的垂直平分线. 2.旋转思考层次 (1)全等变换:对应边相等、对应角相等. (2)对应点与旋转中心 旋转会出现等线段共端点(对应点到旋转中心的距离相等); 对应点与旋转中心的连线所夹的角等于旋转角; 对应点所连线段的垂直平分线都经过旋转中心;
旋转会产生圆(圆弧). (3)常见组合搭配 旋转会出现相似的等腰三角形; 旋转60°会出现等边三角形;旋转90°会出现等腰直角三角形; 相似三角形对应点重合时会出现旋转放缩模型. (4)应用,作图(构造) 当题目(背景)中出现等线段共端点时,会考虑补全旋转构 造全等.(常见背景有正方形、等边三角形、等腰三角形)注:读题标注时,往往要弄清楚旋转三要素; 旋转方向不确定需要分类讨论; 常将图形的旋转转化为点、线段的旋转进行操作.(有时 只需保留研究目标即可)
?精讲精练 1.小明用不同的方式来折叠一个边长为8 的正方形纸片ABCD, 折痕MN 分别与边AD,BC 交于点M,N,沿MN 将四边形ABNM 折叠,点A,B 的对应点分别为点A′,B′.他得到了以下结论:①如图1,当点B′落在DC 的中点处时,BN=5. ②如图2,当点B′落在CD 上时,延长NB′交AD 的延长线于 点E,△NEM 为等腰三角形.③如图2,当点B′落在CD 上时,连接BB′,此时BB′=MN,BB′⊥MN.④如图3,先将正方形沿MN 对折,使AB 与DC 重合,再将AB 沿过点A 的直线折叠,使点B′落在MN 上,则∠MAB′=60°.其中正确结论的序号是. 图1 图2 图 3 2.如图,在△ABC 中,∠ACB=90°,点D,E 分别在AC,BC 上,且∠CDE=∠B,将△CDE 沿DE 折叠,点C 恰好落在AB 边上的点F 处.若AC=8,AB=10,则CD 的长为.
解析几何优化计算6大技巧 中学解析几何是将几何图形置于直角坐标系中,用方程的观点来研究曲线,体现了用代数的方法解决几何问题的优越性,但有时运算量过大,或需繁杂的讨论,这些都会影响解题的速度,甚至会中止解题的过程,达到“望题兴叹”的地步.特别是高考过程中,在规定的时间内,保质保量完成解题的任务,计算能力是一个重要的方面.为此,从以下几个方面探索减轻运算量的方法和技巧,合理简化解题过程,优化思维过程.技巧一回归定义,以逸待劳 回归定义的实质是重新审视概念,并用相应的概念解决问题,是一种朴素而又重要的策略和思想方法.圆锥曲线的定义既是有关圆锥曲线问题的出发点,又是新知识、新思维的生长点.对于相关的圆锥曲线中的数学问题,若能根据已知条件,巧妙灵活应用定义,往往能达到化难为易、化繁为简、事半功倍的效果. 【例题】如图,F 1,F 2是椭圆C 1:x 24y 2 =1与双曲线C 2的公共焦点,A ,B 分别是C 1, C 2在第二、四象限的公共点.若四边形AF 1BF 2为矩形,则C 2的离心率是( ) A.2 B.3 C.32 D.62 【解析】由已知,得F 1(-3,0),F 2(3,0),设双曲线C 2的实半轴长为a ,由椭圆及双曲线的定义和已知, 1|+|AF 2|=4,2|-|AF 1|=2a , 1|2+|AF 2|2=12, 解得a 2=2, 故a = 2.所以双曲线C 2的离心率e =32=62 .【答案】D [关键点拨] 本题巧妙运用椭圆和双曲线的定义建立|AF 1 |,|AF 2|的等量关系,从而快速求出双曲线实半轴长a 的值,进而求出双曲线的离心率,大大降低了运算量.
170℃ 四、化学键理论与分子几何构型 1. NO 的生物活性已引起科学家高度重视,它与O 2- 反应,生成A 。在生理pH 条件下, A 的t 1/2= 1~2秒。 (1) 写出A 的可能的Lewis 结构式,标出形式电荷。判断它们的稳定性。 (2) A 与水中的CO 2迅速一对一地结合,试写出此物种可能的路易斯结构式,表示 出形式电荷,判断其稳定性。 (3) 含Cu +的酶可把NO 2- 转化为NO ,写出此反应方程式。 (4) 在固定器皿中,把NO 压缩到100atm ,发现气体压强迅速降至略小于原压强的 2/3,写出反应方程式,并解释为什么最后的气体总压略小于原压的2/3。 2. 试画出N 5+离子的Lewis 所有可能结构式,标出形式电荷,讨论各自稳定性,写出各 氮原子之间的键级。你认为N 5+的性质如何?它应在什么溶剂中制得。 3. 在地球的电离层中,可能存在下列离子:ArCl +、OF +、NO +、PS +、SCl +。请你预测 哪一种离子最稳定?哪一种离子最不稳定?说明理由。 4. 硼与氮形成类似苯的化合物,俗称无机苯。它是无色液体,具有芳香性。 (1) 写出其分子式,画出其结构式并标出形式电荷。 (2) 写出无机苯与HCl 发生加成反应的方程式 (3) 无机苯的三甲基取代物遇水会发生水解反应,试判断各种取代物的水解方程式, 并以此判断取代物可能的结构式。 (4) 硼氮化合物可形成二元固体聚合物,指出这种聚合物的可能结构,并说明是否具 有导电性。 (5) 画出Ca 2(B 5O 9)Cl·2H 2O 中聚硼阴离子单元的结构示意图,指明阴离子单元的电 荷与硼的哪种结构式有关。 5. 用VSEPR 理论判断下列物种的中心原子采取何种杂化类型,指出可能的几何构型。 (1)IF 3 (2)ClO 3- (3)AsCl 3(CF 3)2 (4)SnCl 2 (5)TeCl 4 (6)GaF 63 - 6. 试从结构及化学键角度回答下列问题:一氧化碳、二氧化碳、甲醛、甲酸等分子 (1) 画出各分子的立体构型,并标明各原子间成键情况(σ、π、Πm n ) (2) 估计分子中碳—氧键的键长变化规律 7. 近期报导了用二聚三甲基铝[Al(CH 3)3]2 (A)和2, 6—二异丙基苯胺(B)为原料,通过 两步反应,得到一种环铝氮烷的衍生物(D): 第一步:A + 2B === C + 2CH 4 第二步:□C □D + □CH 4(□中填入适当系数)请回答下列问题: (1) 分别写出两步反应配平的化学方程式(A 、B 、C 、D 要用结构简式表示 (2) 写出D 的结构式 (3) 设在第一步反应中,A 与过量B 完全反应,产物中的甲烷又全部挥发,对反应后
分子的几何构型和分子的极性 一、杂化轨道理论 1、概念:同一原子中能量接近的不同轨道,在成键过程中,为了发挥更高的 成键效能,可 以重新组合,进行杂化,成为一组能量相同的新轨道。 2、意义:杂化轨道理论证明了共价键的饱和性和方向性,主要用于解释分子 的空间几何构型。 3、类型: 杂化类型杂化轨道数轨道夹角空间构型实例 sp杂化 2 180 直线形HCN、BeCl2、C2H2、CO2 sp2杂化 3 120 平面三角形BF3、CO32-、C2H4、HCHO sp3杂化 4 109°28' 正四面体CH4、CCl4、SO42-、PO43- 4、说明:NH3也采用sp3杂化,但成键后,存在一个孤电子对,对周围的键 产生排斥,使键角不等于109°28',而为107°;H2O也采用sp3杂化,但成键后,存在2个孤电子对,对周围的键产生排斥,使键角不等于109°28',而为104.5°;因为存在2个孤电子对,对周围的键产生排斥加大,因而键角比NH3的更小。CH3Cl也采用sp3杂化,但成键后,不存在孤电子对,键角依旧为 109°28',但因为C—H键长小于C—Cl键长,分子不再是正四面体,而是四面体。 二、价电子互斥理论
1、概念:分子或离子的几何构型主要决定于与中心原子相关的价电子对之间 的排斥作用。价电子对数由成键电子对数和孤电子对数组成。价电子对的排斥作用 孤电子对与孤电子对的排斥,有成键电子对和孤电子对的排斥,还有成键电子对与 成键电子对的排斥,孤对电子间的排斥被认为大于孤对电子和成键电子对之间的排 斥,后者又大于成键电子对之间的排斥。分子会尽力避免这些排斥来保持稳定。当 排斥不能避免时,整个分子倾向于形成排斥最弱的结构(与理想形状有最小差异的 方式)。分子更倾向于最弱的成键电子对与成键电子对的排斥。配体较多的分子中,电子对间甚至无法保持90°的夹角,因此它们的电子对更倾向于分布在多个 平面上。 2、意义:用于预测和分析分子或离子的空间构型。 3、模型:计算中心原子的价电子对数= 注:氧族元素作配位原子认为不提供电子,σ键数为“0”. 孤电子对数=中心原子的价电子对数-σ键数 价电子对成键对数孤电子对杂化类型电子对构型几何构型实例 2 2 0 sp 直线形直线形CO2 3 3 0 sp2 三角形三角形BF3 3 2 1 sp3 三角形V形SO2 4 4 0 sp3 四面体正四面体CH4 4 3 1 sp3 四面体三角锥NH3 4 2 2 sp3 四面体V形H2O 5 5 0 三角双锥三角双锥PCl5 6 6 0 八面体正八面体SF6
首先要分清scf不收敛和几何构型优化不收敛:scf不收敛指的是自洽场叠代不收敛(什么?没听说过什么叫自洽场?那还是回去学习些量化基础知识再开展计算吧),可以认为是对指定结构的波函数不断优化的过程,是为了找到这个某个指定结构下能量最低的波函数,而几何构型优化是对结构的优化的过程,是为了找到某个指定的组分下能量极小结构(注意,不一定是能量最小结构)。在量子化学计算的几何构型优化中,每一步的几何构型优化都包含的很多次的scf计算。 1、scf不收敛的解决方案。 (1) 可以加大scf的循环次数,默认的循环次数是128次,通过scf=(maxcycle=n)来设置最大循环次数n。建议不要超过512,更多的循换没有必要。 (2) 如果加大循环次数不管用,在分子有对称性的情况下,使用scf=dsymm关键词来强制密度对称,有时可以收敛。另外,此关键词很多时候对"scf is confused”这种错误很管用。 (3) 使用scf=symm关键词,使用的前提同上,有时可以收敛。 (4) 如果(2)(3)两步都不行,可以将对称的分子中的某几个原子的位置微调,使分子丧失对称性。这等效于nosymm关键词,但个人经验,这种方式比nosymm好用的多。 (5) 如果还不行,只能拿出杀手锏了,就是使用qc,但不建议直接使用,而是使用xqc关键词,比如scf=(maxcycle=80,xqc),意思是如果scf正常计算(dc)在80个循环之内不收敛才进行昂贵的qc计算,因为scf不收敛多数在几个优化的过程中出现,无法判断哪一步优化的时候会出现scf不收敛,所以用xqc比纯粹使用qc要省时的多。 (6) 中级用户可以在输入文件的井号“#”开头那一行井号后面加上字母"p"来输出更多的信息,其中就有自洽场叠代的信息,分析原因可能会对采用什么方法提供指导。 (7) 前面有虫子提到一个有用的方案但没说清楚,我这里补充一下:如果用用小基组计算,scf可以收敛,那么保存好检查点文件,换成大基组的时候从检查点文件中读取初始猜测(使用guess=read关键词),有时可以算过去。 (8) 如果你计算中使用了含有弥散函数的基组(比如6-31+G*基组中的“+”,Aug-cc-pVTZ基组中的Aug等),可以先去掉弥散函数(比如例子中的两个基组变为6-31G*和cc-pVTZ),这样很容易收敛,需要的话,换成带有弥散函数的基组时,从检查点文件的读初始猜测。原理与(7)类似,有时候管用。注意:(7)和(8)不适合很大的体系,此时,使用小基组或者不带弥散函数的基组与后来使用的大基组在基组数量上差得太多(如果你本来就遇到了scf收敛问题,而使用这两个方案时,机组数量差距超过512个,多数时候要出问题),反倒容易引起scf不收敛,因为读检查点文件时,对那些多出来的基组的猜测是空的,而直接计算时最起码还有个半经验的计算提供了相对合理的初始猜测。 (9) 上述方法有时可以组合使用,如果经过各种组合处理都不收敛,那么放弃吧,你的分子的电子结构太差了。 2、几何构型优化不收敛的解决方案。引起这个问题的原因比较多,可能也说不全,尽量做到全面。 (1) 如果是很小的分子(10原子以内),初始结构可能离平衡结构比较远,又在输出文件优化的最后一步判断是否收敛的位置(可以通过查找Threshold字段找到,它下面有四个判断项,都是YES才代表优化收敛)看到了“-- Number of steps exceeded, NStep=xxx”,可以通过加大优化的循环次数来解决问题。使用关键词opt=(maxcycle=n). (2) 优化到如果四个收敛标准中前两项早就收敛了,而后两项尤其是第三项不收敛,这需要判断原因。如果每一步优化过程中的能量始终在下降,那么可以继续让他算,超过了最大步数停掉了的话把结构拿出来,重新提交就行了。如果能量忽大忽小出现跳跃,说明遇到了比较平坦的势能面,或者优化的算法不好。此时建议按如下顺序处理: a. 使用opt=maxstep=n来缩小最大步长,即原子移动的距离,n的默认值是30,只能设置整