盐酸度洛西丁的合成工艺改进
- 格式:pdf
- 大小:245.63 KB
- 文档页数:4

盐酸金刚乙胺的合成工艺改进嘿,朋友们!今天咱们来聊聊盐酸金刚乙胺的合成工艺改进这档子事儿。
您想想,合成工艺就好比是做菜的流程,想要做出一道美味可口的菜肴,那流程得精心琢磨不是?盐酸金刚乙胺的合成工艺也是这个理儿。
原来的合成工艺啊,可能就像一辆老旧的自行车,嘎吱嘎吱响,还跑得慢。
咱改进它,就是要给它升级换代,变成一辆风驰电掣的跑车!比如说反应条件吧,温度、压力、时间,这可都得拿捏得死死的。
温度高了,那不成了煮过头的粥,全糊啦;温度低了,又像没煮熟的饭,夹生着呢!压力也是,太大了,就像被压得喘不过气的老牛,累得慌;太小了,又使不上劲儿,事办不成。
再看看原料的选择,这就好比选食材,得精挑细选。
质量不好的原料,就像烂了的蔬菜,能做出好菜吗?那肯定不行!还有反应步骤,得简洁高效。
太繁琐了,就像走迷宫,绕来绕去,把自己都绕晕啦;太简单了,又怕关键步骤没到位,功亏一篑。
改进工艺的过程,那可是一场和各种问题的战斗。
有时候,一个小细节没注意,就像一颗小钉子能扎破大轮胎,全盘皆输。
但咱不怕,一次次尝试,一次次改进,就像勇士不断冲锋。
您说,要是合成工艺改进得好,那能带来多少好处啊!生产效率提高了,成本降低了,产品质量更优了,这不就像给企业插上了腾飞的翅膀吗?咱们的科研人员,那可真是绞尽了脑汁,白天黑夜地琢磨。
他们就像辛勤的园丁,精心呵护着这朵合成工艺之花,期待它绽放出最绚烂的光彩。
总之,盐酸金刚乙胺的合成工艺改进,是一项充满挑战但又意义重大的工作。
只有不断探索,不断创新,才能让这工艺变得更加完美,为咱们的生活带来更多的便利和好处!这难道不值得我们全力以赴吗?。

盐酸多西环素的合成工艺研究介绍在医学领域中,多西环素是一种广泛应用的抗微生物药物,它可以通过盐酸多西环素的合成工艺来生产。
本文将探讨盐酸多西环素的合成工艺,包括原料的选择、反应条件的优化以及分离纯化等方面。
原料选择盐酸多西环素的合成需要选择合适的原料。
以下是盐酸多西环素合成工艺中常用的原料: - 对-二氯苯 - 二甲基甲酰胺 - 氨水 - 酥化法制备的多西环素底物反应条件优化为提高盐酸多西环素的产率和纯度,需要对反应条件进行优化。
以下是一些常见的反应条件优化方法: 1. 温度优化 - 在反应初期,较高的反应温度可以促进底物与试剂的反应速度,提高产率。
- 随着反应进行,适当降低温度可以避免副反应的发生,提高纯度。
2. 反应时间控制 - 控制反应时间可以避免过度反应产生的副产物,同时确保足够的反应时间使得底物完全转化。
- 根据反应的进度和底物的含量,合理调整反应时间。
3. 试剂比例控制 - 控制试剂的摩尔比例可以避免废弃试剂的浪费,提高经济效益。
- 合理计算试剂的用量,确保理论过量以保证底物的完全反应。
4. pH值调控 - 在反应过程中,通过调控反应体系的pH值,可以提高产率和纯度。
- 选择合适的缓冲溶液来调节反应体系的pH值。
反应步骤盐酸多西环素的合成工艺包括多个反应步骤。
以下是盐酸多西环素的合成步骤: 1. 反应物悬浮 1. 将对-二氯苯溶于二甲基甲酰胺中,并加入适量氨水。
2. 搅拌反应物混合物,使其悬浮均匀。
2. 反应 1. 将多西环素底物加入反应体系中。
2. 在适宜的温度下控制反应时间,使底物完全转化为盐酸多西环素。
3. 调控反应体系的pH值,使其适合反应进行。
3. 分离纯化 1. 通过酸碱中和使反应体系达到酸性条件,得到盐酸多西环素。
2. 使用适当的溶剂对反应混合物进行提取,使得盐酸多西环素得以分离。
3. 使用旋转蒸发仪将溶剂蒸发,得到纯净的盐酸多西环素。
优化工艺条件为提高盐酸多西环素的合成效果,还需考虑以下方面: 1. 反应容器的选择 - 选择适合的反应容器,能够耐受反应条件下的高温和腐蚀性物质。

盐酸达泊西汀生产工艺盐酸达泊西汀是一种常用的化学药品,它在医药领域有着广泛的应用。
本文将介绍盐酸达泊西汀的生产工艺。
盐酸达泊西汀的生产工艺主要分为以下几个步骤。
首先,将原料达泊西汀与盐酸溶液进行反应。
这一步骤的目的是将达泊西汀与盐酸结合,形成盐酸达泊西汀。
反应过程中需要注意控制温度和反应时间,以确保反应的完全性和产物的纯度。
接下来,对反应混合物进行过滤和干燥处理。
通过过滤,可以去除反应中产生的杂质和固体物质。
干燥处理可以将产物中的水分去除,提高产物的纯度和稳定性。
然后,对干燥后的产物进行结晶处理。
结晶过程中,可以通过控制温度和溶剂的浓度来促使产物结晶。
结晶后的产物通常具有较高的纯度和良好的结晶形态。
对结晶后的产物进行干燥和粉碎处理。
干燥可以进一步去除残留的水分,提高产物的稳定性和保存期限。
粉碎处理可以将产物研磨成所需的粒度,以满足不同的应用需求。
盐酸达泊西汀的生产工艺中,需要注意的一些关键技术点。
首先,反应温度和反应时间的控制十分重要,这可以确保反应的完全性和产物的纯度。
其次,过滤和干燥处理需要采用适当的方法和设备,以提高产物的纯度和稳定性。
此外,结晶过程中的温度和溶剂浓度的选择也对产物的纯度和结晶形态有着重要影响。
盐酸达泊西汀的生产工艺需要严格遵守相关的安全操作规程。
在操作过程中,应注意防止产生有害气体和溶液的泄漏。
同时,应配备必要的防护设施,如安全眼镜、手套和防护服等,以确保操作人员的安全。
盐酸达泊西汀的生产工艺是一个复杂而严谨的过程。
通过控制反应条件、过滤干燥和结晶处理等关键步骤,可以获得高纯度和良好结晶形态的盐酸达泊西汀产物。
在实际生产中,应严格遵守相关的操作规程和安全要求,确保产品质量和操作人员的安全。

收稿日期:2010-09-29作者简介:尹玲丽(1985-),女(汉族),浙江台州人,硕士研究生,E m a i:l y i nli ng li @163.co m;*通讯作者:陈国华(1963-),男(汉族),福建莆田人,副研究员,硕士生导师,主要从事药物化学研究,T e:l (025)83241246,E m a i:l cgh63@163.co m 。
文章编号:1005-0108(2011)01-0037-03盐酸达泊西汀的合成工艺研究尹玲丽,陈国华*(中国药科大学药物化学教研室,江苏南京210009)摘 要:目的研究选择性5 羟色胺重摄取抑制剂盐酸达泊西汀的合成工艺。
方法以3 苯基丙醇和1 氟萘为起始原料,经醚化、溴代、二甲胺基取代、拆分、成盐反应制得目标化合物。
结果与结论目标化合物的结构经1H NM R 、M S 、I R 谱以及比旋光度确证。
该路线原料易得,操作简便,条件温和,有利于工业化生产,消旋体收率达61 2%。
关键词:选择性5 羟色胺重摄取抑制剂;盐酸达泊西汀;化学合成;工艺改进中图分类号:R 914 文献标志码:A盐酸达泊西汀(dapoxetine hydrochlori d e ,1)化学名为(S ) (+) (N,N 二甲胺基) 3 (萘基 1 氧基) 1 苯基丙烷盐酸盐,是一种选择性5 羟色胺重摄取抑制剂(SSRI),由美国礼来制药公司(E li L ill y )研制,2009年在欧洲上市,商品名为Prili g y ,用于治疗男性早泄(PE)。
该药半衰期短、不良反应小、效果显著,是世界上第一种被批准治疗PE 的经口给药的处方药[1]。
1 合成路线文献报道的合成盐酸达泊西汀(1)的方法较多[2-7]。
本研究参考文献[2]的方法,以3 苯丙醇(2)、1 氟萘(3)为起始原料,在氢化钠作用下成醚得到化合物4,4经NBS 苄位溴代得到5,化合物5与二甲胺进行亲核取代反应得到化合物6,6经L (+) 酒石酸拆分,成盐酸盐得目标化合物1。

盐酸多西环素的合成工艺研究
盐酸多西环素是一种广泛应用于医药领域的抗生素,其合成工艺的研
究对于提高其产量和质量具有重要意义。
本文将介绍盐酸多西环素的
合成工艺研究。
首先,盐酸多西环素的合成工艺需要经过多个步骤。
其中,最关键的
步骤是利用四氢呋喃和三氯乙酰氯对二甲基氨基苯酚进行保护,然后
与苯甲酸进行缩合反应,得到N-苯甲酰-2-二甲基氨基苯酚。
接着,
将其与氢氧化钠和氯化亚铁反应,得到N-苯甲酰-2-氨基苯酚。
最后,将其与四环素进行缩合反应,得到盐酸多西环素。
其次,盐酸多西环素的合成工艺需要注意以下几点。
首先,反应条件
需要严格控制,以保证反应的高效性和产物的纯度。
其次,反应中需
要使用高纯度的试剂和溶剂,以避免杂质的产生。
最后,反应中需要
进行多次的分离和纯化,以得到高纯度的产物。
最后,盐酸多西环素的合成工艺研究对于提高其产量和质量具有重要
意义。
通过对反应条件和反应机理的深入研究,可以优化反应条件,
提高反应效率和产物纯度。
同时,可以开发新的合成方法,以提高产
量和降低成本。
此外,还可以对盐酸多西环素的结构和性质进行深入
研究,以拓展其在医药领域的应用。
综上所述,盐酸多西环素的合成工艺研究具有重要意义。
通过对反应条件和反应机理的深入研究,可以优化反应条件,提高产量和质量。
同时,还可以开发新的合成方法,以拓展其在医药领域的应用。

盐酸托莫西汀原料药制备工艺研究摘要:盐酸托莫西汀(atomoxetine,1)的化学名是R-(-)-N-甲基-3-苯基-3-(2-甲基苯氧基)丙胺盐酸盐,为Lilly公司开发,2003年1月在美国上市[1]。
托莫西汀的合成路线主要有两条:Molly路线以3-(N,N-二甲基)氨基-1-苯丙酮为原料,经B2H6还原、SoCl2氯化、Mitsunobu反应、N-去甲基化等4步反应合成目标产物;但没有考虑托莫西汀的旋光性问题,收率不高,反应最后一步需要进行N-去甲基化反应,所用的去甲基化试剂毒性大,反应收率低,且胺基醇的氯代也会有脱水产物的生成。
Ashok路线以苯甲酰基乙酸乙酯为原料,采用化学酶催化的不对称合成,经6步反应得到光学纯度的目标产物,总反应收率较高,只是原料价格较高本文研究了合成药物盐酸托莫西汀的路线。
通过以价廉的苯乙酮为原料,经6步反应制得托莫西汀。
结果目标化合物经1H-NMR、13C-HMR、MS和IR确证,总收率为6.8%。
结论本路线试剂便宜,反应条件温和,操作简单,适合工业化生产。
关键词:抗抑郁药;托莫西汀;合成一、合成路线笔者结合有机合成的理论,在追求低成本、高收率、清洁生产的基础上,参考有关文献,设计了合成路线(图1)。
图1盐酸托莫西汀的合成路线二、仪器与试剂熔点采用WRS-1B数字熔点测定仪测定;红外光谱采用BrokerIFS-55红外光谱仪测定,溴化钾压片;质谱采用ShimadzuGCMS-QP5050A气相质谱联用仪和FinniganSSQ-710型质谱仪测定;核磁共振氢谱、碳谱采用BrzkerARX-300和Varianmercury-300核磁共振仪测定,四甲基硅烷(TMS)为内标;比旋光度用P-E241MC分光旋光仪测定;含量采用岛津SHL-MADCU仪(进样泵:LC10AT,检测器:SPD-10A)测定。
除苯乙酮为工业级外,其他试剂为化学纯。
三、方法和结果3.1N-甲基-β-苯甲酰乙胺盐酸盐的制备将甲胺盐酸盐(16.28g,0.24mol)、多聚甲醛(12.0g,0.40mol)和80mL无水乙醇加入到250mL三颈瓶中,滴加5滴浓盐酸,搅拌加热回流1h至溶液澄清,然后滴加苯乙酮2(24g,0.20mol)(1h加完)。


盐酸达泊西汀的合成及工艺优化作者:付丙月张宁张宗磊段崇刚来源:《中国药房》2020年第07期摘要目的:优化盐酸达泊西汀的合成工艺。
方法:采用手性合成的方法,以3-氯苯丙酮为原料,采用(1S,2R)-(-)-1-氨基-2-茚醇为催化剂、硼烷-N,N-二乙基苯胺为还原剂进行不对称还原,然后依次经过与α-萘酚醚化、磺酸酯化、二甲胺取代、HCl成盐反应得到最终产物。
通过核磁共振和质谱技术对合成产物进行表征。
对中间体Ⅰ、中间体Ⅱ、中间体Ⅲ和最终产物的合成反应进行工艺优化。
结果:表征结果显示最终产物为盐酸达泊西汀,纯度为99.8%,收率为58.9%。
与传统的拆分工艺比较,本工艺采用手性合成的方法不需要拆分,收率明显高于文献报道的拆分工艺收率(31.9%)。
优化后的工艺减少了杂质的产生,提高了产品质量。
结论:本合成工艺反应条件较温和、合成路线较短、收率较高。
关键词盐酸达泊西汀;手性合成;不对称还原;工艺优化ABSTRACT; ;OBJECTIVE: To optimize the synthesis process of dapoxetine hydrochloride. METHODS: By chiral synthesis, asymmetric reduction was carried out by using 3-chlorophenylacetone as raw material,(1S, 2R) -(-)-1-amino-2-indanol as catalyst, and borane-N, N-diethylaniline (DEANB) as reducing agent. Then,it was reacted with α-naphthol etherification, sulfonation, dimethylamine substitution, and HCl salt formation reaction to obtain the final products. The products were characterized by NMR and MS. The synthesis reaction of intermediate Ⅰ, intermediate Ⅱ, intermediate Ⅲ and the final product were optimized. RESULTS: The final product was dapoxetine hydrochloride with purity of 99.8% and yield of 58.9%. Compared with traditional splitting technology, the chiral synthesis technology of this study did not need splitting, and the yield of the technology was significantly higher than that of splitting technology reported in literature (31.9%). The optimized technology reduced the generation of impurities and improved the product quality. CONCLUSIONS: The improved technology has milder reaction conditions, shorter synthesis route and higher yield.KEYWORDS; ;Dapoxetine hydrochloride; Chiral synthesis; Asymmetric reduction; Technology optimization鹽酸达泊西汀(Dapoxetine hydrochloride)化学名为(S)-(+)-(N,N-二甲胺基)-3-(萘基-1-氧基)-1-苯基丙烷盐酸盐,是一种选择性5-羟色胺再摄取抑制剂(SSRIs),2009年在欧洲上市,之后陆续在多个国家和地区上市,2013年进入中国市场(商品名:必利劲),用于成年男子早泄的按需治疗,是世界上第一个被批准用于治疗早泄的口服药[1]。

盐酸哌罗匹隆的合成工艺改进
潘毅;陈蔚;陶勇
【期刊名称】《精细化工中间体》
【年(卷),期】2008(38)4
【摘要】以顺-六氢邻苯二甲酰亚胺(2)为起始原料,经多步反应合成盐酸哌罗匹隆(1)。
中间体3-(哌嗪-1-基)-1,2-苯并异噻唑(9)的制备采用吡啶做催化剂,收率从文献的56.5%提高到71.4%。
总收率从文献的46.4%提高到60.27%。
合成的盐酸哌罗匹隆经1HNMR、13CNMR、MS确证结构。
该合成工艺原料价廉易得、操作简便、不需过柱分离,适合工业化生产。
【总页数】3页(P31-33)
【关键词】工艺改进;哌罗匹隆;非典型抗精神病药物
【作者】潘毅;陈蔚;陶勇
【作者单位】天津药物研究院
【正文语种】中文
【中图分类】R971.41
【相关文献】
1.盐酸罗匹尼罗合成工艺的改进 [J], 余长泉;杨健;许惠钢
2.盐酸哌罗匹隆片联合盐酸氟西汀胶囊对老年脑卒中患者恢复期抑郁临床疗效及生活质量的影响 [J], 王素红;席永涛
3.重复经颅磁刺激联合盐酸哌罗匹隆治疗精神分裂症患者的临床观察 [J], 庞金宇
4.重复经颅磁刺激联合盐酸哌罗匹隆治疗精神分裂症患者的临床观察 [J], 庞金宇
5.艾司西酞普兰+盐酸哌罗匹隆对精神分裂症阴性症状患者的影响 [J], 魏民
因版权原因,仅展示原文概要,查看原文内容请购买。

盐酸多西环素的合成工艺研究盐酸多西环素是一种广泛应用于医药领域的抗生素,其合成工艺的研究对于提高药物的产率和质量具有重要意义。
本文将探讨盐酸多西环素的合成工艺,以及在合成过程中存在的一些关键问题和解决方案。
盐酸多西环素的合成工艺主要分为以下几个步骤:原料准备、前体合成、盐酸多西环素的合成、结晶纯化和干燥。
首先,合成盐酸多西环素的前体化合物是必不可少的。
一般情况下,前体化合物的合成包括物质的选择、反应条件的优化和反应过程的控制。
其中,物质的选择是关键的一步,需要选择具有良好反应活性和高产率的原料。
此外,还需要考虑原料的成本和可获得性。
在前体合成的过程中,反应条件的优化是确保反应的高效进行的关键。
温度、反应时间和反应物的配比等因素都需要仔细考虑。
合适的反应温度和反应时间可以提高反应的速率和产率,同时避免副反应的发生。
反应物的配比需要合理控制,以确保反应物的完全转化和产物的高纯度。
在盐酸多西环素的合成过程中,需要考虑到反应的选择性和产率。
多西环素的合成通常采用氧化还原反应和环化反应。
氧化还原反应是将多西环素的前体化合物转化为多西环素的关键步骤。
在这个过程中,需要选择合适的氧化剂和还原剂,并且控制好反应条件,以确保反应的高效进行。
环化反应是将多西环素的前体化合物经过环化反应转化为多西环素的关键步骤。
在这个过程中,需要选择合适的酸催化剂和反应条件,以确保环化反应的高效进行。
在合成过程中,结晶纯化是非常重要的一步。
结晶纯化可以提高产物的纯度和产率。
合适的溶剂选择和结晶条件的控制是确保结晶纯化的关键。
合适的溶剂选择可以提高产物的溶解度和结晶速率。
结晶条件的控制包括温度、搅拌速度和结晶时间等因素,需要根据具体情况进行调整。
盐酸多西环素的干燥是确保产物质量的最后一步。
干燥的目的是去除产物中的溶剂和水分,以提高产物的稳定性和质量。
合适的干燥条件和方法可以提高干燥的效率和产物的质量。
在盐酸多西环素的合成工艺研究中,存在一些关键问题需要解决。
盐酸舍曲林的合成工艺改进盐酸舍曲林是美国Pfizer 公司开发的强力和高度选择性5-羟色胺再摄取抑制剂,1990 年12月在英国首先上市用于治疗抑郁症,1992 年美国上市。
后又批准用于治疗恐慌症以及强迫观念和行为、惊恐性障碍(Panic Disorder) 、不管是否伴有恐旷症和外伤后的精神紧张性障碍(PTSD)。
[1,2],1998 年我国卫生部批准辉瑞制药XX公司生产的盐酸舍曲林片[(98)卫药准字J-11 号] 在我国上市。
1 合成方法及比较盐酸舍曲林(1) 的制备工艺很多, 基本以4-(3,4- 二氯苯基)-3,4-二氢-1(2H)-萘酮为中间体制得。
文献[3] 报导了氢化的催化剂采用特殊的含铜镉的催化剂, 它以萘烯胺为原料制成Z-盐酸盐的收率83%,收率91%该工艺路线收率较高, 但是氢化的催化剂采用特殊的含铜镉的催化剂, 制作条件复杂, 环境污染较大。
文献[4]报导了以4-(3,4-二氯苯基)-3,4-二氢-1(2H)-萘酮为原料,加入甲胺/甲醇和雷尼镍直接氢化,得Z-盐酸盐,收率48-51%。
工艺的氢化压力为500psi( 约35kg/cm2), 需要压力设备且有关杂质也较高, 重结晶也难以去掉。
文献⑸ 报导了以4-(3,4-二氯苯基)-3,4-二氢-1(2H)-萘酮、甲基羟胺盐酸为原料合成一氧化物, 其结构有立体选择性,催化氢化成为Z-盐酸盐,收率65%本工艺原料较难获得。
文献⑹ 报导了以a-萘酚和邻二氯苯经付-克芳基化、胺基化、脱酰基等反应制得顺式外消旋舍曲林, 再经D-(-)- 酒石酸拆分后成盐即可制得盐酸舍曲林, 总收率25%,本工艺原料较难获得, 反应难于控制, 对设备的要求较高。
另外, 文献[7,8] 还对各种合成路线进行了比较, 提供了一些合成盐酸舍曲林的建议。
在众多合成文献的基础上, 我们确立了以下合成路线:由a -萘酚经过付克反应制得:4-(3,4-二氯苯基)-3,4-二氢-1(2H)- 萘酮(2), 再加入甲胺, 经TiCl4 制得萘烯胺(3), 用Pd/C 氢化,成盐酸盐后顺反异构体分离,制成Z-盐酸盐(4)(收率68%), 再游离,加入R-酒石酸,成酒石酸盐,再游离,成盐酸盐,得到盐酸舍曲林(1) 。
201310020752 盐酸西替利嗪的合成工艺改进丁峰1,2,王风云1*,雷武1,夏明珠1,朱其军2(1.南京理工大学工业化学研究所,江苏南京210094;2.盐城格瑞茵化工有限公司,江苏盐城224400)摘要:该文旨在优化盐酸西替利嗪的原有合成工艺,优化后的工艺以氯苯和苯甲酰氯为起始原料,经傅克酰基化反应、羰基还原反应、羟基氯代反应得到4-氯二苯氯甲烷、再与N-羟乙基哌嗪及氯乙酸钠的亲核加成反应和酸化成盐5步反应制得目标产物盐酸西替利嗪。
与原有合成路线相比,优化后的合成路线减少了一步反应,降低了原料成本,盐酸西替利嗪的总收率从原来的20.9%提高到24.9%,产物纯度在99%以上。
产物结构经红外光谱、核磁共振波谱及质谱进行了表征。
关键词:盐酸西替利嗪;N-羟乙基哌嗪;抗组胺药物Improved synthesis of Cetirizine HydrochlorideDING feng1,2 , Wang Feng-yun1* , Lei Wu1 , XIA Ming-zhu1 , Zhu Qi-jun2 (1School of Chemistry & Engineering, Nanjing University of Science & Technology, Nanjing210094, Jiangsu, China; 2Green Chem (YanCheng) CO. , Ltd. ,Yancheng224400, Jiangsu, China)Abstract: This novel is aimed at optimizing and improving the synthesis route of CetirizineHydrochloride. The new synthesis route use chlorobenzene and benzoyl chloride as startingmaterials, through Friedel-crafts reaction, carbonyl reductive reaction, chlorination,nucleophilic substitution, obtained the product. Compared with the original synthesis route, thissynthesis route shortened one reaction step, reduced the cost of raw materials, The total yield ofCetirizine Hydrochloride was increased from 20.9% to 24.87%. The structure of product wascharacterized by IR, 1H NMR and MS.Key Words: Cetirizine Hydrochloride; N-(2-Hydroxyethyl) piperazine; anti-histamine medicine盐酸西替利嗪是第二代无镇定作用的抗组胺药物,为长效的具选择性的口服强效抗变态反应药,具有较强的生物活性[1,2]。
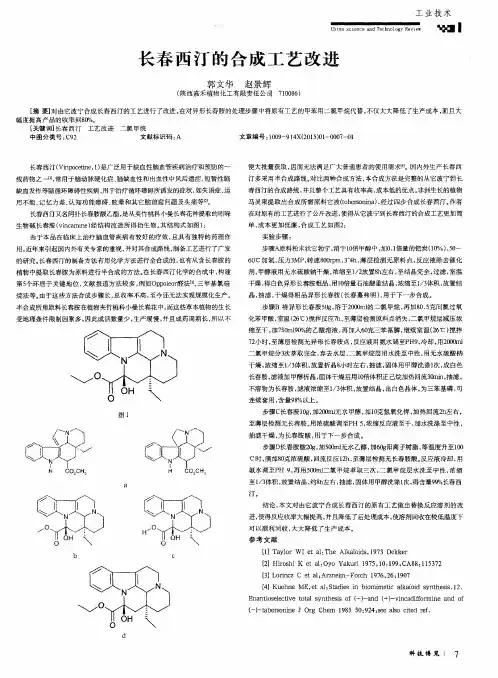
盐酸度洛西汀的合成工艺改进1刘敏,章文军,赵姣河北工业大学化工学院,天津 (300130)Email: liumin_240@摘要:目的本文针对药物盐酸度洛西汀的合成工艺,提出了改进的措施,确立了最佳反应条件,合成了盐酸度洛西汀。
方法以2-乙酰噻吩、二甲胺盐酸盐和多聚甲醛为起始原料,经Mannnich反应、还原、拆分、拆分、醚化、脱甲基、成盐等反应制得盐酸度洛西汀。
对拆分后剩余液中的R构型的N,N-二甲基-3-羟基-(2-噻吩基)-丙胺进行了消旋再拆分,这是主要进行的工艺改进,很适合工业上的再利用理念。
另外更换了部分反应试剂,使得反应收率进一步提高。
结果所得目标化合物的结构经质谱、核磁共振氢谱以及红外光谱确证,总收率由文献报道的21%提高到32%。
结论该工艺可降低成本,操作简化,收率高,适合工业化生产。
关键词:盐酸度洛西汀;合成;消旋再拆分;收率0. 引言盐酸度洛西汀,化学名为 (S)- N- 甲基- 3- (1-萘氧基)- 3-(2- 噻吩基)- 1- 丙胺盐酸盐,是一种对 5- 羟色胺和去甲肾上腺素的再摄取有双重抑制作用的抗抑郁药,它由美国Eli Lilly制药公司开发,商品名为 Cymbalta。
2002 年 9 月经美国 FDA 批准治疗重型抑郁症,临床上用其盐酸盐。
2004 年 9 月,美国 FDA 批准了补充适用证,用于治疗糖尿病性外因神经疼痛[1]。
文献报道了以不同的化合物为起始原料,或选择不同的拆分试剂及脱甲基步序制备盐酸度洛西汀,有多种合成路线,我们选择了以2-乙酰噻吩为起始原料的路线为基本路线,对其进行改进,尤其是在制备(S)-N,N-二甲基-3-羟基-3-(2-噻吩基)丙胺时我们采用了以甲苯为拆分溶剂,并对拆分后剩余的化合物(R)-N,N-二甲基-3-羟基-3-(2-噻吩基)丙胺进行了消旋再拆分工艺使得产率大大的提高。
由于本步的产率提高使得整个路线的产率由文献的21%提高到32%。
合成路线如下:SO3)3)23)21-氟萘3)23HCl31543(S)-32.HCl1本课题得到河北自然科学基金(2005000007)的资助。
1. 试验部分1.1主要仪器与试剂XT-4型双目显微熔点仪(温度计未经校正);Bruker AC-P 400/75型核磁共振仪(以CDCl3:为溶剂。
TMS为内标); Thermo Finnigan LCQ Advantage 貭谱仪。
试验所用试剂均为分析纯;柱层析硅胶 100-200目。
1.2 3-二甲胺基-1-(2-噻吩基)-1-丙酮盐酸盐(2)的制备将28.0g(0.22mol)2-乙酰噻吩、23.3g(0.286mol)二甲胺盐酸盐、13.2g(0.44mol)多聚甲醛和60ml的异丙醇置于250ml的三口瓶中,加入3ml的浓盐酸,加热搅拌回流6h,放入冰水中搅拌1小时,抽滤所得晶体用适量冷乙醇洗涤,滤饼干燥得到白色片状晶体46.3g,收率95.6%,mp.180~181℃(文献[4]:182~184℃)1.3 N,N-二甲基-3-羟基-3-(2-噻吩基)丙胺(3)的制备在250ml三口瓶中加入10g(0.0454mol)的N,N-二甲基-3-羟基-(2-噻吩基)-丙胺溶于120ml甲醇和50ml水中,冷至0℃,加入5mol/L氢氧化钠溶液调至pH11~12,加入硼氢化钠(1.75g,0.0454mol)室温搅拌过夜,TLC监测反应完全,加入15ml丙酮,搅拌半小时,减压蒸去甲醇有白色固体析出,抽滤,50℃真空干燥箱烘干,得消旋体N,N-二甲基-3-羟基-(2-噻吩基)-丙胺粗品,乙酸乙酯精制得7.3g,收率86.4%,m.p.72~73℃(文献[2]71~73℃,收率83%)1.4 (S)-N,N-二甲基-3-羟基-3-(2-噻吩基)丙胺[(S)-3]的制备在250ml三口瓶中加入N,N-二甲基-3-羟基-(2-噻吩基)-丙胺(14g,0.076mol)溶于80ml甲苯中,升温至80℃左右搅拌溶解。
慢慢滴加(S)-(+)-扁桃酸(5.2g,0.034mol)的乙醇(15ml)溶液,反应液透明,滴完后加热回流45分钟,然后冷却至室温搅拌1小时,有白色固体析出,抽滤,固体用少量乙酸乙酯洗涤,然后将乙酸乙酯减压蒸除,得到固体溶于滤液中。
将洗后的滤饼干燥得扁桃酸盐11.12g。
在滤液中加入150ml的稀盐酸(含25g浓盐酸),于室温下反应5h,然后加入25ml5mol/L的氢氧化钠溶液,搅拌后分层,用乙酸乙酯萃取水层。
减压蒸除乙酸乙酯,得到固体合并到甲苯层中。
将上述甲苯溶液搅拌加热到80℃,滴加3g(0.0196mol)(S)-扁桃酸/8ml乙醇溶液,于80℃左右搅拌45min,然后再慢慢冷却至室温搅拌1h,过滤得固体,固体用少量乙酸乙酯洗涤,烘干后得白色固体4.26g。
在滤液中加入80ml稀盐酸(含13.00g浓盐酸),室温下反应5h,然后加入12ml 5mol/L的氢氧化钠水溶液,分层,用乙酸乙酯萃取水层,除乙酸乙酯后的固体和并到甲苯层中。
然后重复处理上述甲苯溶液,得扁桃酸盐 1.86g。
将三次得到的固体共17.24g溶于一定量水中,搅拌下滴加5mol/L氢氧化钠水溶液至溶液pH值11~12,用乙酸乙脂(50ml×3)萃取有机相,用无水硫酸镁干燥,过滤,滤液减压蒸去溶剂,剩余物干燥,得白色固体(S)-3,9.4g,含量达97.6%,收率为68%,[α]D25= -7.6。
,e.e98.6%, (文献[2]收率42.8% ,e.e95%)1.5 (S)-N,N-二甲基-3-(1-萘氧基)-3-2-噻吩基丙胺(4)的制备(S)-N,N-二甲基-3-羟基-(2-噻吩基)丙胺(6g,0.032mol)溶于DMSO(30ml),慢慢分批加入60%氢化钠(1.7g,0.04mol)搅拌0.5h,加入苯甲酸钾(1.1g,7mmol),搅拌10min 后加入1-氟萘(4.4ml,0.042mol),升温至65℃左右反应6h,反应完毕后,将反应液倒入120ml冰水中,加乙酸调pH值至4~5,石油醚(30×3)洗涤,水相加入5mol/L氢氧化钠溶液调至pH11~12,用乙酸乙酯(100ml×3)萃取,有机相拥无水硫酸镁干燥,过滤,滤液家压蒸除溶剂,得红棕色油状物9.7g收率97.2%,[α]D25=+56。
(c 1.0,MeOH)。
1.6 (S)-N-甲基-3-(1-萘氧基)-3-2-噻吩基丙胺(5)的制备9.4g(0.03mol)的(S)-N,N-二甲基-3-(1-萘氧基)-2-噻吩基丙胺溶于100ml甲苯中,然后加热到55℃左右。
加入0.4g(0.003mol)而异丙基乙基胺,搅拌10 min,然后慢慢滴加入4ml 氯甲酸苯酯(0.032mol),温度保持在55℃左右搅拌反应4.5h,TLC检测反应完毕,Rf=0.30(二氯甲烷:甲醇=12:1)然后加入200ml1%NaHCO3,搅拌10min,分层,有机相依次用0.5mol/L 盐酸(50×4),1%NaHCO3溶液(200ml),和水(200ml)洗涤,蒸除甲苯,剩余红棕色油状物,加入DMSO(100ml),将溶液加热至65℃,滴加入10g氢氧化钠/15ml水的溶液,65℃下搅拌反应18h,将反应液倾入100ml冰水中,用冰醋酸调pH=5~5.5。
加入100ml正己烷,搅拌10min,分层。
水相加入5mol/L氢氧化钠溶液调pH值到11~12,然后用乙酸乙酯萃取(100ml×3),有机相拥饱和氯化钠溶液洗(50ml×2),用无水硫酸镁干燥,过滤,将草酸(3.78g,0.03mol)的甲醇(30ml)溶液在室温下加入滤液中,析出大量固体,搅拌0.5h,低温放置过夜,抽滤,乙酸乙酯洗涤滤饼,真空干燥得白色固体度洛西汀草酸盐。
mp:148-151℃。
将度洛西汀草酸盐放入烧杯中加入水(50ml)乙酸乙酯(30ml)用浓氨水(约20ml)中和至固体完全溶解。
分层,乙酸乙酯萃取水层,合并酯层,饱和食盐水洗涤一次,无水硫酸镁干燥,过滤,滤液减压蒸除溶剂,剩余物干燥,得琥珀油7.94g,收率89% 。
R(KBr)υcm-1:3318,3053,2948,2817,2767,1628,1595,1462,1398,1265,1236,1095,1065,771;1H-NMR(CDCl)8.35(1H,m,16-CH),7.78(1H,m,13-CH),7.50(2H,m,14-CH,15-CH),7.38(1H,3d,11-CH),7.26(1H,m,7-CH),7.20(1H,dd,10-CH),7.07(1H,d,5-CH),6.87(2H,m,6-CH,9-CH),5.81(1 H,m,3-CH)2.83(2H,m,2-CH2)2.48(4H,m,-CH3,-NH),2.25(2H,t,1-CH2);EMI-MS(m/s):297.8(M+1)298.9(M+2).1.7 (S)-N-甲基-3-(1-萘氧基)-3-2-噻吩基丙胺盐酸盐(1)的制备将5g(S)-N-甲基-3-(1-萘氧基)-3-2-噻吩基丙胺溶于80ml乙酸乙酯中,冰盐浴,温度控制在-1℃~0℃,通入HCl气体,不断有粘稠的黄棕色固体析出,至沉淀完全,抽滤,用乙酸乙酯洗涤,然后加100ml丙酮回流搅拌,此时固体颗粒变为细小的白色针状结晶,过滤后干燥得3.6g产品,产率为65.4%,mp:164-165℃, [α]D25=+87。
(c 1.0,MeOH)。
2. 结论在制备2时,该反应为典型的曼尼希反应,文献收率较高(73%)。
按照文献[4]反应溶剂为乙醇,但得到的产物曼尼希碱盐酸盐在乙醇中溶解度较大,造成一定的损失。
因此,改用异丙醇做溶剂,提高了收率。
经过大量的试验分析确定了,该步反应最佳的反应条件:2-乙酰噻吩的浓度为0.4mol/L,2-乙酰噻吩:二甲胺盐酸盐:多聚甲醛投料摩尔比为1:1.3:2,反应时间为6h,使收率提高到95.6%。
在制备3时,对溶剂进行了改进:将原来的溶剂乙醇换成了甲醇:水=12:5,可使2-噻吩-2-二甲胺基乙酮盐酸盐全部溶解还原进行彻底产物产率提高,同时也省去了后处理中的酸化碱化过程,使收率达到86.4%。
在拆分3时,选用甲苯为拆分溶剂,一.是因为(S)-3-扁桃酸盐在甲苯中的溶解度较低,以快速成盐析出,水解后收率达到47%。
二.是甲苯易回收价格低廉并且可作为下步醚化反应的溶剂直接使用。
并且本实验对化合物(R)-3进行了消旋再拆分,参照文献[8]的方法,在甲苯和稀盐酸混合溶液中成功将化合物(R)-3 消旋。
经消旋再拆分,总的还原拆分收率由文献[2]报道的42%提高到68%,由于本步产率的大大提高,从而大大提高了总的产率,使总产率达到32%。