枸橼酸西地那非原料药新的未知杂质的发现与合成
- 格式:pdf
- 大小:1.89 MB
- 文档页数:4

2018年第26卷 第6期,450 ~45+合成化学Chinese Journal of Synthetic ChemistryVol.26, 2018No.6, 450 -453•制药技术•抗抑郁药盐酸度洛西汀的合成及其中间体杂质的研究季程宇1,谢童杰1 !吴佳佳1 !马良秀2 !郝之奎1 !颜剑波2 !徐峰!(1.台州职业技术学院化学制药研究所,浙江台州318000! 2.浙江新东港药业股份有限公司,浙江台州318000)摘要:以乙酰噻吩和甲胺盐酸盐为原料,经M am ch缩合、酶法还原和缩合共3步反应合成了抗抑郁药盐酸度洛 西汀,总收率50.6%,并对第一步反应中的3个杂质进行了iH NMR,13C N M R和M S(ESI)分析,确证为1,5-二(噻吩-2-基)戊烷二酮,3-乙氧基-1%噻吩-2-基)-1-丙酮和(,(二[(噻吩-2-基)丙酮% -基]甲胺。
并对反应机理 进行了探讨。
关键词:乙酰噻吩;度洛西汀;酶法;杂质;合成中图分类号:R914.5 文献标志码:A D O I:10. 15952/j. cnki. cjsc. 1005-1511.2018.06. 17219 Study onthe Synthesis of AntidepressionDrug Duloxetine Hydrochloride and Its Impurity of IntermediatesJI Cheng-yu1,XIE Tong乙e1,WUJia乙a1,MA Liang-xiu2 ,HAOZhi-kui1,YANJian乙o2,XUFeng1*(1. Chemical Pharmaceutical Research Institute,Taizhou Vocational & Technical College,Taizhou 318000,China; 2. Neo-Dankong Pharmaceutical C o.,L td.,Taizhou 318000,China) Abstract:Duloxetine hydrochloride,w itli the total yield 50.6%,was prepared by a three-step reaction of Mannich condensation,enzymatic reduction and condensation from acetyltliiophene and metliyl-amine hydrochloride.Three impurities in the first step was analyzed by1H NMR, 13C NMR and MS(E S I),and their structures were confirmed as 1,5-d i(thiophen-2-y l)pentane-1,5-dione,3-ethoxy-1-(thiophen-2-y l)propan-1-one and 3,3’-(methylazanediyl)bis(1-( thiophen-2-y l)propan-1-one).The plausible reaction mechanism was discussed as well.Keywords:acetylthiophene;Duloxetine;enzymatic method;impurity;synthesis度洛西汀(Duloxetine)是由美国E lililly公司 开发的一种5-羟色胺和去甲肾上腺素双重再吸 收抑制剂(S N R I),较其他抗抑郁药(如帕罗西 汀、氟西汀和瑞波西汀等)具有更好的安全性和 耐受性,更少的不良反应,以及多样的治疗活性 (可用于糖尿病)[1]。

厄贝沙坦的合成新工艺研究现代社会,人们生活节奏加快,生活工作压力越来越大,加上饮食作息不规律,造成高血压已经成为高发的心脑血管疾病,患病数量呈逐年上升的态势,患病年龄也在逐年降低,严重影响工作与生活的质量。
因此,除了改善作息规律,加强身体锻炼,按照医生建议进行药物治疗,能达到立竿见影的效果。
对抗高血压的药物市面上常见西药有六个大的品类,厄贝沙坦就属于其中的血管紧张素Ⅱ受体拮抗剂类药物,是降压效果好、副作用少的常规用药。
厄贝沙坦1997年就上市了,最先在欧洲和美国上市。
最主要的功能就是治疗成年人的原发性的高血压,也可以与其他药物进行配伍,作为降压药物治疗方案,治疗高血压和2型糖尿病成年人患者的肾病。
目前在沙坦类药物中,厄贝沙坦的销量排在第二位,具有广阔的市场空间。
因此,为了降低成本,减少三废的产生,对厄贝沙坦新的合成工艺进行研究,具有非常大的经济效益和环境效益。
在过往的文献中,主流的合成工艺,是以环戊酮为起始原料,环戊酮与氰化钠、氯化铵反应生成1-氨基环戊腈草酸盐,反应得到2-丁基-1,3-二氮杂螺环-[4,4]-壬-1-烯-4-酮中间体,再与2-氰基-4’-溴甲基联苯之间发生缩合反应得到的中间体,再用叠氮钠反应,来构建四唑环,得到厄贝沙坦。
这些报道过的合成方法,都用到氰化物,对环境和个人都非常不利。
本研究的目的是设计一条全新的合成路线,避免采用剧毒氰化物。
课题启动时,设计的合成路线是把甘氨酸作为起始的原材料,通过与正戊酰氯反应,在与4-溴苄胺反应生成双酰胺的中间体,然后在酸的催化下,脱水缩合构建氮杂环,再与1,4-卤代丁烷发生合环反应,生成螺环产物2-丁基-3-(4’-溴苯甲基)-1,3二氮杂螺[4,4]壬-1-烯-4-酮。
其再与2-[N-(三苯甲基)-四氮唑]苯硼酸发生SUZUKI偶联生成带保护基的厄贝沙坦中间体,酸性条件下脱掉四唑环上的保护基团,得到厄贝沙坦。
但在合成研究过程中在得到双酰胺化合物后,无法得到脱水缩合产物。

吡唑醚菌酯原药的主要杂质及合成研究高士杰;樊凯;张金凯;管阳凡;郑锦彪;胡晓明【摘要】There were three impurities with relatively high contents in pyraclostrobin TC during the synthesis process. The impurities were prepared from pyraclostrobin TC and their structures were characterized by NMR and MS. The standard material of pyraclostrobin was also prepared through recrystallization. The study provided support for the synthesis process of pyraclostrobin TC and full components analysis.%吡唑醚菌酯原药中含有3个在生产过程产生的含量较高的杂质,以吡唑醚菌酯原药为原料,合成了这些杂质,NMR和MS表征了其结构,并通过对原药重结晶纯化制得标准品,为吡唑醚菌酯原药合成工艺的控制和全组分分析提供支持.【期刊名称】《现代农药》【年(卷),期】2017(016)006【总页数】5页(P17-21)【关键词】吡唑醚菌酯;杂质;标准品;合成【作者】高士杰;樊凯;张金凯;管阳凡;郑锦彪;胡晓明【作者单位】上海晓明检测技术服务有限公司,上海 200335;上海晓明检测技术服务有限公司,上海 200335;上海晓明检测技术服务有限公司,上海 200335;上海晓明检测技术服务有限公司,上海 200335;上海晓明检测技术服务有限公司,上海200335;上海晓明检测技术服务有限公司,上海 200335【正文语种】中文【中图分类】TQ450.1吡唑醚菌酯(pyraclostrobin)又名唑菌胺酯,是含有吡唑结构的新型甲氧基丙烯酸酯类杀菌剂。

西格列汀的合成温度西格列汀(Sildenafil)是一种用于治疗男性勃起功能障碍(ED)的药物。
它是一种磷酸酯酶-5(PDE5)抑制剂,通过抑制PDE5酶的作用来增加阴茎海绵体内一氧化氮(NO)的浓度,从而促进血管扩张,增加血流量,帮助男性获得并维持足够的勃起。
西格列汀的合成温度是指在制备过程中所需的合适温度。
合成温度的选择对于药物的质量和产量都有重要影响。
下面将介绍西格列汀的合成温度及其制备过程。
西格列汀的合成温度通常在常温下进行。
制备过程中的温度控制非常重要,过高或过低的温度都可能导致反应的不完全或副反应的发生。
西格列汀的制备过程相对较为复杂,主要包括以下几个步骤:1. 原料准备:西格列汀的合成需要苯甲酸、氯乙酸、硝酸银等原料。
这些原料需要经过精细处理和准备,以确保反应的顺利进行。
2. 反应步骤:首先将苯甲酸与氯乙酸缩合生成苯甲酸乙酯,然后与硝酸银反应,生成硝酸苯甲酯。
接下来,将硝酸苯甲酯与硝酸甲酯进行酯交换反应,生成硝酸甲酯苯甲酯。
最后,将硝酸甲酯苯甲酯与乙醇胺反应,生成西格列汀。
3. 温度控制:在上述反应过程中,温度的控制非常重要。
一般来说,反应温度在常温下进行,通常在20-30摄氏度之间。
过高的温度可能会导致反应的不完全,而过低的温度则可能使反应速率过慢。
4. 反应时间:除了温度控制外,反应时间也是制备西格列汀的关键因素之一。
反应时间的长短直接影响到产率和纯度。
一般来说,反应时间在数小时至数天之间。
5. 结晶和纯化:反应结束后,需要对产物进行结晶和纯化。
这一步骤可以帮助去除杂质,提高纯度。
结晶温度的选择也需要谨慎,一般来说,在适宜的溶剂中加热溶解,然后慢慢冷却至室温,即可得到结晶的西格列汀。
总结起来,西格列汀的合成温度通常在常温下进行,控制温度的合适与否对于产物的质量和产率都有重要影响。
在制备过程中,除了温度的控制外,反应时间和结晶纯化也是需要注意的关键因素。
通过合理控制这些条件,可以获得高纯度的西格列汀药物,从而提高治疗效果。

康奈非尼原料药杂质合成工艺目录一、基本信息 (3)1.1药品名称 (3)1.2结构 (3)二、康奈非尼杂质列表 (4)三、杂质合成工艺 (6)3.1杂质A的合成 (6)3.2杂质B的合成 (6)3.3杂质C的合成 (7)3.4杂质D的合成 (7)3.5杂质E的合成 (7)3.6杂质F的合成 (8)3.7杂质G的合成 (8)3.8杂质H的合成 (9)3.9杂质I的合成 (9)康奈非尼原料药杂质合成工艺一、基本信息1.1药品名称中文通用名:康奈非尼英文通用名:Encorafenib汉语拼音:KANGNAIFEINI中文化学名:(3S)-N-[5-[(2R)-2-(2,5-二氟苯基)-1-吡咯烷基]吡唑并[1,5-A]嘧啶-3-基]-3-羟基-1-吡咯烷甲酰胺英文化学名:(3S)-N-[5-[(2R)-2-(2,5-Difluorophenyl)-1-pyrrolidinyl]pyrazolo[1,5-a]pyrimidin-3-yl]-3-hydroxy-1-pyrrolidinecarboxamide化学文摘登记(CAS)号:1223403-58-4外观与性状:白色至类白色固体上市时间:2018年06月27日1.2结构结构式:分子式:C22H27ClFN7O4S分子量:540.01立体结构:含1个手性中心作用机理:BRAFTOVI康奈非尼(Encorafenib)与BINIMETINIB联合用于治疗患有BRAFV600E或V600K突变的不可切除或转移性黑色素瘤患者。
苏黎世大学医院皮肤癌中心Dummer等报告表示,Encorafenib与维罗非尼(Vemurafenib)相比,无论是其联合Binimetinib或者它用于单药治疗都显示出了优于维罗非尼的好疗效。
不过,Encorafenib 与Binimetinib联合用药比Encorafenib单药治疗或维罗非尼有更好的耐受性。
康奈非尼合成路线二、康奈非尼杂质列表三、杂质合成工艺3.1杂质A的合成合成操作:取质量为10g的SM1、8g的SM2-1化合物加入至250ml高压反应釜中,加入异丙醇40ml溶解。
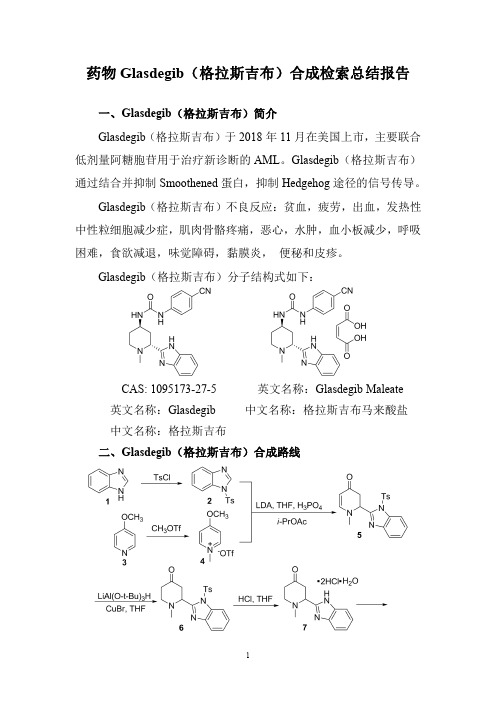
药物Glasdegib(格拉斯吉布)合成检索总结报告一、Glasdegib(格拉斯吉布)简介Glasdegib(格拉斯吉布)于2018年11月在美国上市,主要联合低剂量阿糖胞苷用于治疗新诊断的AML。
Glasdegib(格拉斯吉布)通过结合并抑制Smoothened蛋白,抑制Hedgehog途径的信号传导。
Glasdegib(格拉斯吉布)不良反应:贫血,疲劳,出血,发热性中性粒细胞减少症,肌肉骨骼疼痛,恶心,水肿,血小板减少,呼吸困难,食欲减退,味觉障碍,黏膜炎,便秘和皮疹。
Glasdegib(格拉斯吉布)分子结构式如下:CAS:1095173-27-5英文名称:Glasdegib Maleate英文名称:Glasdegib中文名称:格拉斯吉布马来酸盐中文名称:格拉斯吉布二、Glasdegib(格拉斯吉布)合成路线三、Glasdegib (格拉斯吉布)合成检索总结报告(一)Glasdegib (格拉斯吉布)中间体2的合成合成方法实验步骤参考文献合成方法一To a three-necked flask with an overhead stirrer was charged benzimidazole 1(118g,1.00mol)and EtOAc (1.20L).The solution was cooled to 10°C,TsCl (191g,1.00mol)was added in one portion followed by Et 3N (139mL,1.00mol).The resultant slurry was then aged at 20°C for four hours.The reaction was considered complete when <2%of benzimidazole was observed by UPLC analysis.To the slurry was added 300mL of water and the layers were separated.The organic phase was washed with 2x 300mL of water.The organic was then concentrated under vacuum to a volume of ~500mL,2.00L of isopropanol was added,and then concentrated again under vacuum to final volume of ~1.00L and a slurry was formed.The slurry was cooled to 20°C and water (1.00L)was added over 30minutes.The slurry was aged at 20°C for two hours and filtered,washed with 2x 500mL of water,and dried in an oven under vacuum at 60°C for 24hours to give 2as white solids (248g,99.0wt%,90.0%corrected yield).Organic Letters ;vol.16;nb.3;(2014);p.860–863.(二)Glasdegib (格拉斯吉布)中间体4的合成合成方法实验步骤参考文献合成方法一To athree-necked flask was charged 4-methoxypyridine 3(102mL,1.00mol)and CH 2Cl 2(550mL).The solution was cooled to 0°C,and MeOTf (113mL,1.00mL)was added slowly,keeping the internal temperature below 20°C.The solution was then aged at 20°C for one hour.The solvent was concentrated under vacuum and switched to MTBE until a final volume of ~2.00L.The resultant slurry in MTBE was aged at 20°C for one hour,filtered,and the solids were washed with MTBE,and dried in an oven under vacuum at 40°C for six hours to give 4as white solids (270g,99.0%yield):m.p.65–66°anic Letters ;vol.16;nb.3;(2014);p.860–863.(三)Glasdegib (格拉斯吉布)中间体5的合成合成方法实验步骤参考文献合成方法一To a three-necked flask equipped with an overhead stirrer was charged 2(100g,366mmol)and THF (1.00L)under nitrogen.The solution was cooled to -20°C.A solution of LDA (2M in THF/heptane/ethylbenzene,201mL,403mmol)was added slowly,keeping internal temperature <-10°C.After the addition,the dark-brown solution was aged at -20to -10°C for 10minutes.A solution of 4(100g,366mmol)in THF (500mL)was added to the above solution while keeping the internal temperature <-10°C.The resultant slurry was then aged at -20to -10°C for 10minutes.The slurry was then quenched into a cold (0°C)aqueous H 3PO 4solution (2M,403mL,806mmol)while maintaining the internal temperature <10°C and pH <5.The resultant solution was aged at 10°C for one hour and UPLC analysis showed <1%of the enol ether intermediate.To the mixture was then added an aqueous solution of KOH (2N,~604mL)until pH >8.0was achieved.Isopropyl acetate (500mL)was then added and the layers wereOrganic Letters ;vol.16;nb.3;(2014);p.860–863.。
收稿日期:2019-02-07作者简介:李淑恒(1987—),女,回族,甘肃张家川,硕士研究生,专业方向:有机合成。
枸橼酸西地那非原料药新的未知杂质的发现与合成李淑恒1,宋煜伟2,申鹏翠1,董静静1,潘会歌1(1.新乡制药股份有限公司药物研究所,河南新乡 453000;2.洛阳职业技术学院食品药品学院,河南洛阳 471000)摘要:枸橼酸西地那非原料药合成过程中出现的新的未知杂质进行了提纯、结构表征及合成。
将氯气通入5-(2-乙氧苯基)-1-甲基-3-丙基-1,6-二氢-7H-吡唑[4,3-d]嘧啶--酮的氯仿溶液中合成杂质-(-氯--乙氧苯基)-1-甲基-3-丙基-1,6-二氢-7H-吡唑[4,3-d]嘧啶-7-酮,并经过LS-MS、NMR、IR确证结构。
此杂质未见报道,文献调研及实验研究发现该杂质是造成枸橼酸西地那非原料药纯度降低的主要因素之一,为确保药物安全有效,合成过程中严格控制该杂质的含量。
本研究论文侧重于对该杂质形成机理及结构进行表征分析。
关键词:枸橼酸西地那非;杂质;表征;合成中图分类号:R979.9 文献标识码:A 文章编号:1008-021X(2019)08-0054-04TheDiscoverydndSynthesisofNewUnknownImpurityinCitrateSildenaLiShuheng1,SongYuwei2,ShengPengcui1,DongJingjing1,PanHuige1(1.XinxiangPharmaceuticalCo.,Ltd.,InstituteofPharmacology,Xinxiang 453000,China;2.LuoyangPolytechnic,CollegeofFoodandPharmaceuticalSciences,Luoyang 471000,China)Abstract:Thenewunknownimpurityappearedintheprocessofthesynthesisofsildenacitratewaspurified,characterizedandsynthesized.Thechlorinegasbubbledintochloroformsolutionof5-(2-ethoxyphenyl)-1-methyl-3-propyl-1,6-dihydro-7H-pyrazolo[4,3-d]pyrimidin-7-onetosynthesis5-(5-chloro-2-ethoxyphenyl)-1-methyl-3-propyl-1,6-dihydro-7H-pyrazolo[4,3-d]pyrimidin-7-one,andthestructurewereconfirmedbyMS、NMR、IR.Thisimpuritywasnotreportedthatliteratureresearchesandexperimentalstudiesfoundthattheimpurityisoneofthemainfactortoreducethepurityofsildenafilcitrate.inordertoensurethedrugissafeandeffective,strictcontroloftheimpuritycontentinthesyntheticprocessshouldbetakenseriously.Thispaperfocusesonthecharacterizationoftheformationmechanismandstructureoftheimpurity.Keywords:sildenafilcitrate;impurity;characterization;synthesis 枸橼酸西地那非药品标准已经列入欧洲药典(EP),美国药典(USP)和印度药典(IP),目前明确了的有关物质2有:合成工艺控制中残留的3个有关物质,分别为1-甲基-3-丙基-4-氨基吡唑-5-甲酰胺(盐酸盐),1-甲基-3-丙基-4-(2-乙氧基苯甲酰基)吡唑-5-甲酰胺(Ⅰ),1-甲基-3-丙基-5-(2-乙氧基苯基)-6,7-二氢-1H-吡唑并[4,3-d]嘧啶-7-酮(Ⅱ)。
药典规定的其他有关物质有:5-[(2-乙氧基-5-(4-甲基哌嗪-1-磺酰基)苯基]-1-甲基-3-(2-甲基丙基)-1,6-二氢吡唑并[4,3-d]嘧啶-7-酮(Ⅲ),3-乙氧基-3-(1-甲基-7-氧代-3-丙基-6,7-二氢-1H-吡唑并[4,4-d]嘧啶-5-基)苯磺酸(Ⅳ),1-[(4-乙氧基-3-[5-(6,7-二氢-1-甲基-7-氧代-3-丙基-1H-吡唑并[4,3-d]嘧啶)]苯磺酸基〗-4-甲基哌嗪-4-氧化物(Ⅴ),5-[(2-羟基-5-(4-甲基哌嗪-1-磺酰基)苯基]-1-甲基-3-丙基-1,6-二氢吡唑并[4,3-d]嘧啶-7-酮(Ⅵ),结构式详见图1。
在我公司枸橼酸西地那非的实验中,高效液相色谱检测枸橼酸西地那非过程中,存在一个未知杂质(以下均简称Imp2-1),此杂质非图1所示的有关物质,并且作为枸橼酸西地那非原料药的最大单杂存在,高于药典规定的单杂控制限度。
因此对Imp2-1的研究以此展开,确保药物安全有效,对该杂质的定性与含量控制应受到重视。
1 实验1.1 仪器与试剂Waters2695高效液相色谱仪,Waters2695质谱联用仪及empower数据处理工作站(美国沃特世科技有限公司);核磁共振仪brukerDPX-40M(德国布鲁克科技有限公司);perkinelmerspectrumtwo红外光谱仪(铂金埃尔默公司);旋转蒸发器(郑州长城科工贸有限公司);循环水式真空泵(郑州长城科工贸有限公司);干燥箱(上海博迅实业有限公司医疗设备厂)。
图1 有关物质的结果Fig.1 structuresofrelatedsubstance 1-甲基-3-正丙基-4-氨基吡唑-5-甲酰胺盐酸盐(盐酸盐)、氯磺酸、N-甲基哌嗪、乙酸乙酯、三乙胺、氢氧化钾、无水乙醇、碳酸氢钠、一水合柠檬酸、二氯甲烷、色谱级乙腈、色谱级甲醇、去离子超纯水。
1.2 枸橼酸西地那非原料药的合成工艺枸橼酸西地那非原料药的合成方法报道较多[3-11]。
方法大同小异,必需原料与反应路径相同。
一般的合成方法如图2:(1)将盐酸盐、乙酸乙酯,三乙胺投入反应瓶,10℃以下慢慢滴加邻乙氧基苯甲酰氯,抽滤干燥得Ⅰ。
(2)将Ⅰ、氢氧化钾和无水乙醇投入三颈烧瓶回流,反应液降至室温盐酸调节pH值至,抽滤干燥得Ⅱ。
(3)将氯磺酸滴加入反应瓶,10℃以下分批缓慢加入Ⅱ,30℃反应。
反应结束后,将反应液缓慢倒入冰水和二氯甲烷的混合液中搅拌分层,有机相Ⅳ收入反应瓶中,滴加N-甲基哌嗪,反应液用饱和碳酸氢钠溶液、去离子超纯化水分别萃取一次,有机相减压蒸馏得西地那非。
酸,乙醇,西地那非至三颈烧瓶中回流,降温结晶抽滤,得枸橼酸西地那非粗品。
枸橼酸西地那非粗品经活性炭,%乙醇回流精制,得枸橼酸西地那非成品。
图2 枸橼酸西地那非的合成路线Fig.2 SyntheticRouteofsildenafilcitrate1.3 Imp2-1的研究在该合成工艺下,枸橼酸西地那非成品中检测出杂质Imp2-1,液相百分比高于0.1%。
在本研究发现Imp2-1之前,依据欧洲药典中所列的枸橼酸西地那非有关物质检测项下的HPLC条件,在该HPLC条件下,Imp2-1保留时间靠后而未被检测出,为此本研究制定了一个针对Im2-1的HPLC条件。
经过HPLC保留时间对比,Imp2-1在中间体Ⅱ中已经存在(含0.2%左右,见图3),经过后续第3步反应,生成产物Ⅳ的二氯甲烷萃取液中,Imp2-1在薄层色谱上清晰可见。
在第3步反应的基础上,Ⅱ在改变氯磺酸用量以及反应温度的体系中,所得产物Ⅳ中,Imp2-1的液相百分比可提升至15%,接着对Ⅳ进行质谱分析,得Imp2-1分子质量为346.9。
由于第2步与第步反应产物都存在Imp2-1,而两步反应唯一相同的元素为体系里都含有氯化氢(氯磺酸分解产生H2SO4和HCl)。
最终推测出Imp2-1的结构可能为5-(5-氯-2-乙氧苯基)-1-甲基-3-丙基-1,6-二氢-7H-吡唑[4,3-d]嘧啶-7-酮。
图3 Imp2-1和中间体ⅡHPLC峰综合报告Fig.3 PeakComprehensiveReportofHPLCofImp2-1andⅡ2 Imp2-1的合成及结构表征2.1 Imp2-1的合成具体的合成方法如图4:将10gⅡ、100mL氯仿投入三口瓶1中,加入1mL氯磺酸搅拌均匀;10g高锰酸钾投入三颈烧瓶2中,恒压滴液漏斗中加入60mL盐酸装与三颈烧瓶2,三颈烧瓶1与三颈烧瓶2之间连起通气管,通气管口应没入氯仿液面以下,设备准备完毕,开启盐酸滴注,观察三口瓶1的气泡,确定盐酸滴注速度,同时开启搅拌并加热回流三颈烧瓶1。
当不再有氯气气泡产生时停止反应。
用水萃取,减压蒸馏有机相,乙酸乙酯重结晶,干燥得产物1g,液相纯度达97%以上。
通过液相保留时间与质谱对比,确认得到的产物为5-(5-氯-2-乙氧苯基)-1-甲基-3-丙基-1,6-二氢-7H-吡唑[4,3-d]嘧啶-7-酮。
在没有氯磺酸存在的体系中,直接通入氯气,并不能得到Imp2-1。
图4 Imp2-1的合成路线Fig.4 SyntheticRouteofImp2-12.2 杂质的结构表征Imp2-1的NMR见表1,IRspectra见图5,LS-MS见图6。
表1 Imp2-1的核磁数据Tab.1 NMRofImp2-1图5 Imp2-1的红外图谱Fig.5 IRspectraofImp2-1图6 Imp2-1的液相质谱Fig.6 LS-MSSpectrumofImp2-12.3 Imp2-1的普遍性在对Imp2-1研究的同时,我们也购入了其他公司生产的枸橼酸西地那非,其他厂家的产品也同样存在Imp2-1(图7,还有其他厂家的样品同样含有Imp2-1,由于大量图谱的名称以代码形式显示不具有代表性而未在此图列出)。
因为HPLC进样方法采用点样,枸橼酸西地那非主峰高不同,但是已经超出了药典规定的有关物质检测浓度,不影响有关物质及Imp2-1的含量检测。
图中可见,其他厂家的Imp2-1明显存在,可见大部分生产厂家也是在本文列出的工艺下生产的。
本公司实验室小试产品为枸XDNF2#,通过工艺改进(工艺涉嫌机密,无法列出),已将Imp2-1彻底除去,并将Imp2-1列入了枸橼酸西地那非成品的质量标准中加以控制。
图7 枸橼酸西地那非HPLC峰综合报告Fig.7 PeakCombrehensiveReportofHPLCsildenafilcitrate3 总结Imp2-1未见报道,研究表明Imp2-1是由体系中存在HCl而产生的,而氯磺酸是工艺中必不可少的原料,氯磺酸自身在其磺化反应过程中会产生大量HCl。