微卫星筛选方法课件24页PPT文档
- 格式:ppt
- 大小:282.50 KB
- 文档页数:24
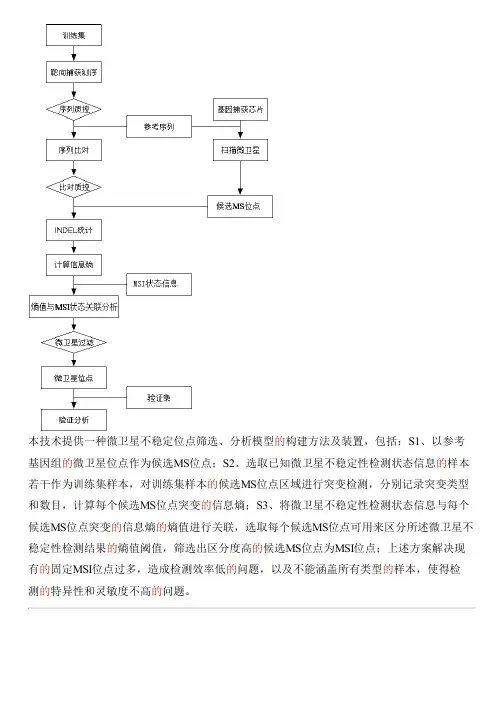
本技术提供一种微卫星不稳定位点筛选、分析模型的构建方法及装置,包括:S1、以参考基因组的微卫星位点作为候选MS位点;S2、选取已知微卫星不稳定性检测状态信息的样本若干作为训练集样本,对训练集样本的候选MS位点区域进行突变检测,分别记录突变类型和数目,计算每个候选MS位点突变的信息熵;S3、将微卫星不稳定性检测状态信息与每个候选MS位点突变的信息熵的熵值进行关联,选取每个候选MS位点可用来区分所述微卫星不稳定性检测结果的熵值阈值,筛选出区分度高的候选MS位点为MSI位点;上述方案解决现有的固定MSI位点过多,造成检测效率低的问题,以及不能涵盖所有类型的样本,使得检测的特异性和灵敏度不高的问题。
权利要求书1.一种微卫星不稳定位点筛选模型的构建方法,其特征在于,包括:S1、以参考基因组的微卫星位点作为候选MS位点;S2、选取已知微卫星不稳定性状态信息为阳性和阴性的样本若干作为训练集样本,对所述训练集样本的候选MS位点区域进行突变检测,分别记录突变类型和数目,计算每个所述候选MS位点突变的信息熵;所述信息熵的计算公式如下式(1):(1)其中,H(Xi)代表所述候选MS位点的熵值;代表每个所述候选MS位点的Indel数目,;代表每个所述候选MS位点的Indel数目占所有候选MS位点Indel数目总和的百分比,;S3、将所述微卫星不稳定性状态信息与每个所述候选MS位点突变的信息熵的熵值进行关联,选取每个所述候选MS位点可用来区分微卫星不稳定性状态的熵值阈值,选择筛选出前k 个区分度最高的候选MS位点为MSI位点,k≤候选MS位点总数目,所筛选的候选MS位点的熵值阈值区分所述训练集样本的微卫星不稳定性状态的假阳性比率和假阴性比率均<5%。
2.根据权利要求1所述的微卫星不稳定位点筛选模型的构建方法,其特征在于,在S2步骤中,在突变检测前,还包括对用于突变检测的测序数据进行质控,将质控后的测序数据与参考基因组比对,并对比对结果文件的捕获效率、目标区域平均测序深度、微卫星位点的覆盖深度和污染率进行阈值筛选,选出阈值范围内的候选MS位点进行后续突变检测。

高效快速筛选新发现的微卫星位点的方法-HRM摘要:本文介绍了一种新的识别微卫星位点的方法,微卫星标记是一种功能强大的共显性标记,扩增产物可靠且可以确定已知物种的高度的多态信息含量,但是对于新位点的发现依然是一种费时费力的方法。
相对于大肠杆菌文库分离方法,新一代测序技术(NGS)可以得到更多可用的候选基因。
我们对已知微卫星引物的注释作了系统的论述,并发现无论采用什么分离技术,由于PCR 未充分扩增,基因位点的单态性或多拷贝都会导致一半以上的候选基因丢失。
因此,在分子标记技术上,筛选候选基因依然是一个很关键的步骤。
我们通过重新评估毛细电泳法进行基因分型,发现HRM可以用于基因分型及筛选候选基因,对此列出了具体的流程。
通过该流程,或许我们可以快速地进行新一代标记的检测,并可以把费用降低到传统方法的一半甚至四分之一。
关键词:HRM 微卫星重新分离技术分子标记新一代测序技术群体遗传学引言微卫星也叫简单重复序列(SSRs),是群体遗传学中最强大的共显性标记,具有广泛的应用(e.g., Chambers and MacAvoy 2000; Ellegren 2004; Jarne and Lagoda 1996;MacDonald et al. 2011)。
最近由于新一代测序技术(NGS)的发展正解决SSRs技术中的一个重要的缺陷,不能获得足够的设计引物的数据。
扩增片段多态位点的选择,依然是一件费时费力的工作,这是SSRs发展中的另一个瓶颈。
因此我们引进了HRM用于识别潜在的SSR位点,并且HRM有望加快SSR的发展速度,降低传统方法的费用。
SSRs 一般是以核心序列1-6bp为重复单位首尾串联而成的短的DNA序列(Wan et al. 2004)。
微卫星具有高突变率、易于进行PCR扩增、便于毛细电泳法(CE)的分析以及可以利用许多软件工具进行生物学推论的诸多优点,使其在群体遗传学方面得到广泛应用。
传统的SSR位点的重新分离,是借助于丰富的基因文库(Glenn and Schable 2005; Zane et al. 2002),但是该法对于SSR位点不多的物种则不能检测到足够的位点进行分析(Arthofer et al. 2007; Megle´cz et al. 2004)。

微卫星DNA简单重复序(SSR)也称微卫星DNA,也可称为SSRP(Simple Sequence Repeat Polymorphisms),STMS (Sequence-tagged microsatellites)。
其串联重复的核心序列为1一6 bp,其中最常见是双核昔酸重复,即(CA) n和(TG) n每个微卫星DNA的核心序列结构相同,重复单位数目10一60个,其高度多态性主要来源于串联数目的不同。
SSR标记的基本原理:根据微卫星序列两端互补序列设计引物,通过PCR反应扩增微卫星片段,由于核心序列串联重复数目不同,因而能够用PCR的方法扩增出不同长度的PCR产物,将扩增产物进行凝胶电泳,根据分离片段的大小决定基因型并计算等位基因频率。
SSR具有以下一些优点:(l)一般检测到的是一个单一的多等位基因位点;(2)微卫星呈共显性遗传,故可鉴别杂合子和纯合子;(3)所需DNA量少。
显然,在采用SSR技术分析微卫星DNA多态性时必须知道重复序列两端的DNA序列的信息。
如不能直接从DNA数据库查寻则首先必须对其进行测序。
1 微卫星DNA的构成及特点微卫星又称简单序列重复(Simple Se-quence Repeats,SSR)。
一般以1-6个碱基为核心序列,首尾相连组成的串联重复序列。
这种序列存在于几乎所有真核生物的基因组中,含量丰富,且呈随机均匀分布。
它们不仅大量分布于基因的间隔区和内含子中,而且还分布于基因的外显子和调控区(如启动子、增强子),真核生物平均每50-150 kb就存在一个微卫星位点,如在人类基因组中每6 kb就有1个微卫星,禽类基因组中约89 kb出现1个微卫星。
微卫星DNA 数目巨大,人类基因组中约有5×104-1×105个(CA)n重复序列,重复次数一般为15-60次,重复单位相同,其长度一般小于200 bp,但也有的更长。
每个特定位点的微卫星DNA均由两部分构成:中间的核心区和外围的侧翼区。



凡纳滨对虾微卫星序列的筛选马宁;曾地刚【摘要】为了筛选凡纳滨对虾微卫星序列,构建了一个cDNA文库,并进行454高通量测序.结果得到500 177条mRNA序列片段,通过拼接获得了20 225条unigene,从中鉴定得到588条微卫星序列,设计了557对微卫星引物.随机挑选其中20对微卫星引物进行PCR扩增,结果有18对引物扩增成功.研究结果大大丰富了凡纳滨对虾的基因组资料,增加了凡纳滨对虾可用的微卫星标记数量,对于凡纳滨对虾的种质资源鉴定、遗传学研究和分子标记选育有重要意义.【期刊名称】《西南农业学报》【年(卷),期】2013(026)006【总页数】5页(P2629-2633)【关键词】凡纳滨对虾;微卫星序列;高通量测序【作者】马宁;曾地刚【作者单位】广西水产研究所遗传育种与健康养殖重点实验室,广西南宁530021;广西水产研究所遗传育种与健康养殖重点实验室,广西南宁530021【正文语种】中文【中图分类】S945.1微卫星(Microsatellites)又被称为简单重复序列(Simple sequence repeats,SSRs),由DNA上的2~6个核苷酸的串联重复片段构成[1],它们在真核生物的整个基因组中均匀分布。
微卫星标记由于重复片段的重复次数在个体间呈高度多态,共显性遗传,所以被广泛应用在动植物的分子标记遗传育种研究中[2~3]。
微卫星位点多态性的检测方法通常是根据微卫星两旁的DNA序列,设计引物进行PCR扩增,然后通过聚丙烯酰胺凝胶电泳或毛细管电泳,根据PCR产物的分子量差别进行检测。
以往筛选微卫星标记的方法,主要先构建微卫星富集文库,然后随机挑选克隆进行测序,需要耗费大量的时间、人力和资金。
高通量测序是近年来新发展的一种DNA测序技术,它是在芯片上大规模并行地对数百万计的DNA分子同时进行测序,一次测序可以产生巨大数据量的测序结果。
近年来,由于高通量测序技术的发展,使我们能够大规模、高效率和低成本的对物种的基因组进行测序。

1.进行酶切用Vs pⅠ对四种鱼(GC、GS、NL和ZZ)基因组DNA(需基因组DNA浓度较高)进行酶切。
反应体系为:ddH2O 15µLVs pⅠbuffer 2µLVs pⅠ酶1µLDNA 2µL总体积20µL酶切在37℃反应3~4h即可,但是为了提高酶切效率可切4~6h。
PCR反应程序为:94℃ 5min,(94℃ 1min,53℃ 1min,72℃ 1min)×30,72℃ 10min,4℃ hold。
Vs pⅠ酶即(AseⅠ酶):酶切识别位点为:5’A T↓T A A T 3’5’T A A T↑T A 3’AseⅠ酶的接头为:A1:5’CTC GTA GAC TGC GTA CC 3’A2:5’TAG GTA CGC AGT C 3’AseⅠ酶用的预扩增产物为:A3:5’CTC GTA GAC TGC GTA CCT AAT 3’2.接头制备用10µL A1(200µM)和10µL A2(200µM)混合,94℃ 3min后室温放置30min。
3.酶切片段与接头连接接头为A1和A2制备的接头,命名为AE。
连接反应体系如下:ddH2O 5µLT4 ligation酶0.5µLT4 buffer4µL酶切产物20µLA TP(10mM)0.3µLAE 1µL总体积30.8µL上述混合体系在16℃连接过夜。
10h左右。
4.连接产物PCR扩增对连接产物进行PCR扩增,每个样本做4管。
扩增引物为A3。
反应体系如下:ddH2O 14µL10×buffer 2.5µLMg2+2µLdNTPs(10mM)0.5µLA3 1µLDNA(连接产物)5µLTaqE(2.5U/µL)0.2µL总体积25.2µLPCR反应程序为:(94℃ 30sec,56℃ 1min,72℃ 1min)×20,72℃ 10min,4℃ hold。
微卫星DNA简单重复序(SSR)也称微卫星DNA,也可称为SSRP(Simple Sequence Repeat Polymorphisms),STMS (Sequence-tagged microsatellites)。
其串联重复的核心序列为1一6 bp,其中最常见是双核昔酸重复,即(CA) n和(TG) n每个微卫星DNA的核心序列结构相同,重复单位数目10一60个,其高度多态性主要来源于串联数目的不同。
SSR标记的基本原理:根据微卫星序列两端互补序列设计引物,通过PCR反应扩增微卫星片段,由于核心序列串联重复数目不同,因而能够用PCR的方法扩增出不同长度的PCR产物,将扩增产物进行凝胶电泳,根据分离片段的大小决定基因型并计算等位基因频率。
SSR具有以下一些优点:(l)一般检测到的是一个单一的多等位基因位点;(2)微卫星呈共显性遗传,故可鉴别杂合子和纯合子;(3)所需DNA量少。
显然,在采用SSR技术分析微卫星DNA多态性时必须知道重复序列两端的DNA序列的信息。
如不能直接从DNA数据库查寻则首先必须对其进行测序。
1 微卫星DNA的构成及特点微卫星又称简单序列重复(Simple Se-quence Repeats,SSR)。
一般以1-6个碱基为核心序列,首尾相连组成的串联重复序列。
这种序列存在于几乎所有真核生物的基因组中,含量丰富,且呈随机均匀分布。
它们不仅大量分布于基因的间隔区和内含子中,而且还分布于基因的外显子和调控区(如启动子、增强子),真核生物平均每50-150 kb就存在一个微卫星位点,如在人类基因组中每6 kb就有1个微卫星,禽类基因组中约89 kb出现1个微卫星。
微卫星DNA 数目巨大,人类基因组中约有5×104-1×105个(CA)n重复序列,重复次数一般为15-60次,重复单位相同,其长度一般小于200 bp,但也有的更长。
每个特定位点的微卫星DNA均由两部分构成:中间的核心区和外围的侧翼区。

1.进行酶切用Vs pⅠ对四种鱼(GC、GS、NL和ZZ)基因组DNA(需基因组DN A浓度较高)进行酶切。
反应体系为:d dH2O15µLVs pⅠbuffer 2µLVs pⅠ酶1µLDNA2µL总体积20µL酶切在37℃反应3~4h即可,但是为了提高酶切效率可切4~6h。
PCR反应程序为:94℃ 5min,(94℃ 1min,53℃ 1min,72℃ 1min)×30,72℃ 10min,4℃ hold。
Vs pⅠ酶即(AseⅠ酶):酶切识别位点为:5’A T↓T A A T 3’5’T A A T↑T A 3’AseⅠ酶的接头为:A1:5’CTC GTA GAC TGC GTA CC 3’A2:5’TAG GTA CGC AGT C 3’AseⅠ酶用的预扩增产物为:A3:5’CTC GTA GAC TGC GTA CCT AAT 3’2.接头制备用10µLA1(200µM)和10µLA2(200µM)混合,94℃ 3min后室温放置30min。
3.酶切片段与接头连接接头为A1和A2制备的接头,命名为AE。
连接反应体系如下:ddH2O5µLT4 ligati on酶0.5µLT4 buffer4µL酶切产物20µLA TP(10mM)0.3µLAE 1µL总体积30.8µL上述混合体系在16℃连接过夜。
10h左右。
4.连接产物PC R扩增对连接产物进行PCR扩增,每个样本做4管。
扩增引物为A3。
反应体系如下:ddH2O14µL10×buffer 2.5µLMg2+2µLdNTPs(10mM)0.5µLA3 1µLDNA(连接产物)5µLTaqE(2.5U/µL)0.2µL总体积25.2µLPCR反应程序为:(94℃ 30sec,56℃ 1min,72℃ 1min)×20,72℃ 10min,4℃ hold。

