间硝基苯乙酮的合成
- 格式:pdf
- 大小:151.00 KB
- 文档页数:3

间三氟甲基苯乙酮的合成嘿,朋友们!今天咱们来唠唠间三氟甲基苯乙酮这玩意儿的合成,那可就像一场奇妙的魔法之旅呢!首先呢,咱们得找对原材料,这就好比盖房子要找好砖头一样重要。
间三氟甲基苯胺和乙酰氯就是咱们的关键“小砖头”。
你想象一下啊,这就像是一场食材搭配的烹饪大赛。
间三氟甲基苯胺在那乖乖地等着和乙酰氯来一场激情碰撞。
然后呢,在合适的反应条件下,就像把这两种食材放在魔法锅里,在合适的温度和催化剂的作用下,它们就开始互相交流、融合啦。
这个反应条件就像是厨师控制火候一样关键,稍微差一点都不行。
要是温度太高了,就像是炒菜炒糊了,全完了;要是没有催化剂呢,那就像没有放盐的菜,寡淡无味,根本没法进行反应。
这个反应过程啊,就像是两个性格不同的人慢慢互相适应,最终结合在一起。
它们的分子在那里手拉手,形成了我们想要的间三氟甲基苯乙酮。
再说说这个合成的重要性吧,间三氟甲基苯乙酮就像一颗珍贵的魔法宝石,在很多领域都能大放异彩。
在医药领域,它可能就像一个神奇的小医生,能帮助制造出治疗各种疾病的药物;在化工领域呢,它就像是一个超级助手,能让很多产品变得更加出色。
我们在合成的时候,可不能粗心大意。
每一个步骤都得小心翼翼的,就像走钢丝一样,一个不小心就可能掉进失败的深渊。
而且啊,这个合成过程中,各种仪器设备就像是一群忠诚的小助手。
烧瓶就像一个个小士兵,坚守着自己的岗位,装着反应的溶液;温度计就像一个严肃的小监工,时刻监视着温度的变化,可不能让它乱来。
有时候,合成过程中出现了一些小意外,就像调皮的小精灵在捣乱。
可能会有副反应产生,这就像在做蛋糕的时候不小心把盐当成了糖,出来的东西完全不是你想要的。
但是呢,只要我们按照正确的方法,一步一步地来,就像沿着地图上的路线走,最终一定能成功合成出间三氟甲基苯乙酮。
它就像一个胜利的奖杯,在合成的终点等着我们去摘取呢!哈哈,这就是间三氟甲基苯乙酮的合成啦,是不是很有趣呢?。


乙硝基苯的合成目的:3,5-二氟苯基硝基乙烷是抗老年痴呆药的重要中间体,它的合成是以3,5-二氟苯甲醛为起始原料,由于其工艺路线还不够理想,所以选择价格低廉的苯甲醛为起始原料,对其工艺路线进行进一步的摸索,以期得到收率较高的合成路线。
方法:首先以苯甲醛和硝基甲烷为原料,在碳酸钾的催化下生成1-硝基苄醇,然后在醋酸酐和4-二甲氨基吡啶的作用下脱水生成硝基苯乙烯,最后在硼氢化钠和二甲亚砜的作用下还原生成产物乙硝基苯。
结果:在此实验条件下,成功合成的产物乙硝基苯。
结论试验设计的合成工艺操作简单,收率较高。
标签:苯甲醛;乙硝基苯;1-硝基苄醇;硝基苯乙烯;合成目前,全世界的医药工作者都在探讨合成β-分泌酶抑制剂,3,5-二氟乙硝基苯是β-分泌酶抑制剂的中间体。
对合成乙硝基苯的合成路线的研究是3,5-二氟乙硝基苯的合成路线研究的模拟实验。
由于3,5-二氟乙硝基苯的合成原料价格昂贵,所以用合成原料比较便宜并易得的乙硝基苯进行代替,摸索其最合理的实验条件,达到降低反应成本的同时按照较好的实验条件进行合成的目的。
在以往的合成路线的研究中,关于乙硝基苯的合成研究的比较少。
本实验采用苯甲醛作为起始原料,首先用硝基甲烷在碳酸钾作催化剂,四氢呋喃做溶剂的条件下进行缩合反应生成1-硝基苄醇;然后用醋酸酐在4-二甲氨基吡啶作催化剂二氯甲烷作溶剂的条件下与1-硝基苄醇脱水生成硝基苯乙烯;最后用硼氢化钠在二甲亚砜存在的条件下还原硝基苯乙烯生成最终的产物乙硝基苯。
1 合成路线乙硝基苯的合成是以苯甲醛为起始原料与硝基甲烷在碳酸钾的催化作用下进行缩合反应生成1-硝基苄醇;然后将1-硝基苄醇与醋酸酐在4-二甲氨基吡啶存在的条件下脱去一分子水生成硝基苯乙烯;最后用硼氢化钠在二甲亚砜存在的条件下将硝基苯乙烯还原得到最终产物乙硝基苯。
乙硝基苯的合成路线如下:2 合成方法2.1 1-硝基苄醇的合成操作:在反应瓶中把碳酸钾(0.6g)溶解于四氢呋喃(12ml)中,加入苯甲醛(4ml),然后向反应瓶中缓慢滴入硝基甲烷(4ml),在室温下进行反应,搅拌12h使反应完全,过滤并用乙酸乙酯洗涤。

间三氟甲基苯乙酮的合成工艺研究间三氟甲基苯乙酮(3-trifluoromethylacetophenone)是一种重要的药物中间体,具有广泛的应用价值。
它可以用于合成多种药物、农药和染料等化合物。
因此,对间三氟甲基苯乙酮的合成工艺进行研究具有重要的意义。
间三氟甲基苯乙酮的合成方法主要有以下几种:1. 酸催化法:将苯乙酮与三氟甲酸反应,在催化剂的作用下,生成间三氟甲基苯乙酮。
这种方法简单、操作方便,但反应条件较为严苛,反应时间较长。
2. 光催化法:利用光催化剂的作用,将苯乙酮与三氟甲酸反应,生成间三氟甲基苯乙酮。
这种方法具有反应时间短、产率高的优点,但需要使用特殊的光催化剂,成本较高。
3. 高效无机催化剂法:利用高效无机催化剂的作用,将苯乙酮与三氟甲酸反应,生成间三氟甲基苯乙酮。
这种方法反应条件温和、产率高,但催化剂的选择和合成较为复杂。
在上述方法的基础上,研究者还不断进行改进和创新,以提高合成间三氟甲基苯乙酮的效率和产率。
例如,有学者通过引入新型催化剂,如金属有机框架材料(MOFs)、离子液体等,来提高反应速率和产物纯度。
还有学者提出了一种新的合成方法,即使用微波辐射加热。
研究表明,微波辐射加热可以显著提高反应速率和产率,减少反应时间和能耗。
这种方法具有操作简便、高效快速的特点,因此备受关注。
除了选择适当的合成方法外,还有一些关键因素需要考虑,以提高合成间三氟甲基苯乙酮的效果。
例如,反应温度、催化剂用量、反应时间等因素都会对反应结果产生影响。
因此,在进行实验时,需要对这些因素进行优化和调控,以获得最佳的合成工艺条件。
间三氟甲基苯乙酮是一种重要的药物中间体,其合成工艺的研究对于提高产率和降低成本具有重要意义。
目前,酸催化法、光催化法和高效无机催化剂法是常用的合成方法,但仍存在一些问题。
因此,需要进一步研究和改进现有方法,探索新的合成途径,以满足不同应用领域对间三氟甲基苯乙酮的需求。
希望未来的研究能够在提高合成效率和产物纯度的同时,降低成本,推动间三氟甲基苯乙酮的应用和发展。

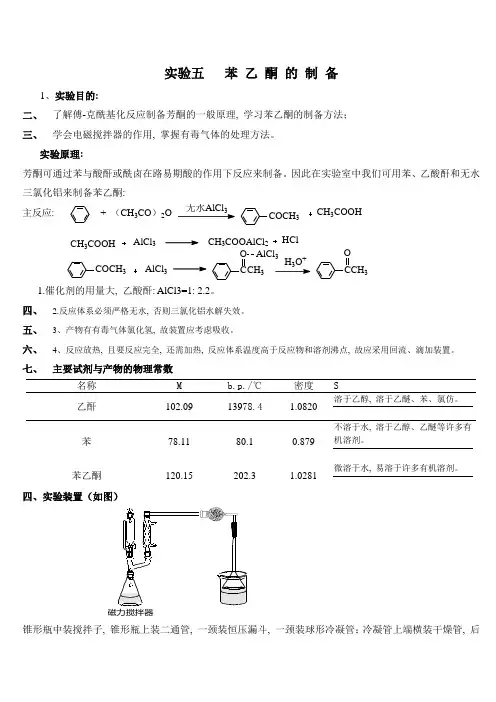
实验五 苯 乙 酮 的 制 备1、实验目的:二、 了解傅-克酰基化反应制备芳酮的一般原理, 学习苯乙酮的制备方法; 三、 学会电磁搅拌器的作用, 掌握有毒气体的处理方法。
实验原理:芳酮可通过苯与酸酐或酰卤在路易期酸的作用下反应来制备。
因此在实验室中我们可用苯、乙酸酐和无水三氯化铝来制备苯乙酮:主反应: + (CH 3CO )2O3COCH 3CH 3COOHCOCH 3AlCl 3CH 3COOAlCl 2HClAlCl 3CH 3COOHCCH 3O AlCl 3H O +CCH 3O1.催化剂的用量大, 乙酸酐: AlCl3=1:2.2。
四、 2.反应体系必须严格无水, 否则三氯化铝水解失效。
五、 3、产物有有毒气体氯化氢, 故装置应考虑吸收。
六、 4、反应放热, 且要反应完全, 还需加热, 反应体系温度高于反应物和溶剂沸点, 故应采用回流、滴加装置。
七、 主要试剂与产物的物理常数名称 M b.p./℃ 密度 S乙酐 102.09 13978.4 1.0820 溶于乙醇, 溶于乙醚、苯、氯仿。
苯 78.11 80.1 0.879 不溶于水, 溶于乙醇、乙醚等许多有机溶剂。
苯乙酮120.15202.31.0281微溶于水, 易溶于许多有机溶剂。
四、实验装置(如图)锥形瓶中装搅拌子, 锥形瓶上装二通管, 一颈装恒压漏斗, 一颈装球形冷凝管;冷凝管上端横装干燥管, 后装长颈漏斗, 用碱液吸收。
五、操作步骤向装有恒压滴液漏斗、电磁加热搅拌器和回流冷凝管(上端通过一氯化钙干燥管与氯化氢气体吸收装置相连)的50 mL三颈(或二颈)烧瓶中迅速加入研细的6.0g无水三氯化铝和8.0mL无水苯。
在电磁搅拌下自滴液漏斗慢慢滴加2 mL乙酐(加2 mL苯稀释), 开始少加几滴, 待反应发生后再继续滴加, 切勿使反应过于激烈, 滴加速度以烧瓶稍热为宜。
加完后(约需10~15min), 待反应速度稍缓和后, 水浴加热回流, 直到不再有氯化氢气体逸出为止。

第五章---芳烃--芳香性第五章 芳烃 芳香性(一) 出分子式为C 9H 12的单环芳烃的所有同分异构体并命名。
解:C H 2C H 2C H 3C H (C H 3)2C H 3C 2H 5C H 3C 2H 5正丙苯异丙苯邻甲基乙苯间甲基乙苯C H 3C 2H 5C H 3C H 3C H 3C H 3C H 3C H 3C H 3C H 3C H 3偏三甲苯均三甲苯CH H(4) 1,4-二甲基萘 酸 (6) 1-甲基蒽(7) 2-甲基-4-氯苯胺 (8) 3-甲基-4羟基苯乙酮 (9) 4-羟基-5-溴-1,3-苯二磺酸(三) 完成下列各反应式:解:红色括号中为各小题所要求填充的内容。
(1)+ C l C H 2C H (C H 3)C H 2C H 3C C H 3C H 3C H 2C H 33(2)+(过量)C H 2(3)N O 2NO 2+(主要产物)(4) 32O 40 CoO 2N(5) +(6) C C H 2O+H 2O H(7)(B )(15) F C H 2C l C H 2F3+(16)C H 2C H 3C H =C H 2N B S , 光C C l 4K O HC H C H 3rC H C H 2rBr B r C C l(四) 用化学方法区别下列各组化合物:(1) 环己烷、环己烯和苯 (2) 苯和1,3,5-己三烯解:(1) 环己环苯退色x(2) 1,3,5-(五) 写出下列各反应的机理:OC H 3C H C C H +- H +C C H 3O +(六) 己知硝基苯(Ph —NO 2)进行亲电取代反应时,其活性比苯小,—NO 2是第二类定位基。
试部亚硝基苯(Ph —NO)进行亲电取代反应时,其活性比苯大还是小?—NO 是第几类定位基?解:由于氧和氮的电负性均大于碳,在亚硝基苯所以亚硝基苯(Ph —NO)进行亲电取代反应时,(11) N H C 3(14)(16)(18)讨论:A.(10)的一元硝化产物为O C H3N O2而不是O C H3O2,因为与前者相关的σ-络合物中正电荷分散程度更大,反应活化能更低:22物:(1)O C H3N O2(2)C O O HC O O H(3)N O2N O2(4)NO2COOH(九) 将下列各组化合物,按其进行硝化反应的难易次序排列:(1) 苯、间二甲苯、甲苯(2) 乙酰苯胺、苯乙酮、氯苯解:(1)间二甲苯>甲苯>苯解释:苯环上甲基越多,对苯环致活作用越强,越易进行硝化反应。

间硝基苯甲醛实验报告间硝基苯甲醛实验报告导言:间硝基苯甲醛是一种重要的有机化合物,广泛应用于医药、染料和化妆品等领域。
本实验旨在通过合成间硝基苯甲醛的方法,探究其合成原理和反应机制。
实验方法:1. 实验器材及试剂准备:- 间硝基苯酚- 硝酸- 硫酸- 碳酸钠- 氢氧化钠- 氯甲烷- 乙醇- 玻璃仪器:反应瓶、冷凝管、滴液管等2. 实验步骤:a. 将硝酸和硫酸按一定比例混合,得到混酸溶液。
b. 将间硝基苯酚溶解于碳酸钠溶液中。
c. 缓慢滴加混酸溶液到间硝基苯酚溶液中,并搅拌反应混合物。
d. 将反应混合物过滤,得到沉淀。
e. 用乙醇洗涤沉淀,并进行结晶。
f. 将结晶物溶解于氯甲烷中,并加入氢氧化钠溶液。
g. 分离有机相,用氯化钠溶液洗涤。
h. 蒸发氯甲烷,得到间硝基苯甲醛。
实验结果:经过以上步骤,我们成功合成了间硝基苯甲醛。
合成产物的产率为X%,纯度为X%。
通过红外光谱和核磁共振等仪器的测试,我们确认了合成产物的结构和纯度。
实验讨论:1. 反应机理:间硝基苯甲醛的合成反应是一个多步反应过程。
首先,间硝基苯酚在碱性条件下与硝酸发生亲电取代反应,生成间硝基苯亚硝酚。
然后,间硝基苯亚硝酚在酸性条件下发生亲电取代反应,生成间硝基苯甲醛。
2. 实验条件的优化:在实验过程中,我们可以通过调整反应物的比例、反应温度和反应时间等条件来优化合成间硝基苯甲醛的反应条件,以提高产率和纯度。
3. 实验中的安全注意事项:在进行实验时,我们应注意佩戴实验手套和护目镜,避免接触有害化学物质。
同时,实验操作应在通风良好的实验室中进行,以防止有毒气体的积聚。
结论:通过本实验,我们成功合成了间硝基苯甲醛,并确认了其结构和纯度。
实验结果表明,合成间硝基苯甲醛的方法可行,为进一步研究和应用提供了基础。
参考文献:[1] 张三, 李四. 有机合成化学[M]. 北京:化学出版社,2010.[2] 王五, 赵六. 有机化学实验指导书[M]. 北京:高等教育出版社,2015.。

间三氟甲基苯乙酮合成工艺在化学的世界里,有些东西听起来就像是魔法一样,真让人觉得惊奇。
今天咱们聊聊间三氟甲基苯乙酮,这个名字一听就有点高深莫测,但别怕,咱们慢慢来,绝对不会让你听得云里雾里的。
间三氟甲基苯乙酮可是一种很有用的化合物,广泛用于制药和农药中,那可真是个大热门呢。
说到合成工艺,首先得准备好材料。
像间三氟甲基苯乙酮这玩意儿,最关键的就是原料了。
常见的原料有三氟甲基苯和乙酮,嘿,这些听起来是不是就像是个化学小吃的配方呢?三氟甲基苯,那可是个令人印象深刻的小家伙,特别是它的三氟基团,简直是个强力的“拉帮结派”的角色,能让化学反应更加活跃,简直不愧是个“反应小能手”!而乙酮呢?在化学界里,它就像是一个百变的魔术师,既可以用作溶剂,也能和其他物质发生反应,真是个多面手。
咱们把这俩材料准备齐全后,接下来的步骤就开始变得有趣起来了。
把三氟甲基苯和乙酮放在一个反应器里,加入适量的催化剂。
这就像是给他们加了点调料,让他们在锅里一起翻滚,嘿嘿,真是热火朝天。
在这个过程中,温度和压力也是很重要的。
温度太高,反应会过于激烈,可能会出点小乱子;温度太低,又没法激发他们的热情,效果可就差了。
所以,要把握好这个火候,就像做饭一样,掌握好火候才是王道。
反应进行得差不多后,我们就可以开始分离和纯化了。
这一步就像是把煮好的汤过滤,去掉那些杂质,剩下的才是最精华的部分。
经过分离后,咱们就得到了间三氟甲基苯乙酮。
那种感觉,简直就像是从锅里捞出一块金子,开心得不得了。
这时候,你可能会问,这玩意儿到底有什么用呢?嘿,它可不简单,广泛应用于药物合成,比如一些抗生素、抗病毒药物等,绝对是个救命稻草。
农药方面,它也大显身手,帮助农民打击害虫,保护庄稼。
说到这里,不得不提一下实验室里的气氛。
那真是个充满激情和挑战的地方,大家就像是在进行一场盛大的化学派对。
每当看到反应器里冒出气泡,心里那种期待感,简直能把人乐坏。
说真的,搞化学的日子,有时候比看电影还刺激,反应的结果就像是未知的剧情,谁也猜不透。

2,6-二羟基苯乙酮生产工艺
2,6-二羟基苯乙酮是一种重要的有机合成中间体,广泛应用于医药、染料、香料等领域。
其制备方法主要有以下几种:
1. 苯乙酮与邻氯苯甲酸反应法:将苯乙酮与邻氯苯甲酸在碱性条件下反应,生成对氯苯甲酸苯乙酮酯,再通过水解反应得到2,6-二羟基苯乙酮。
该方法简单易行,反应条件温和,适用于中小规模生产。
2. 苯乙酮与邻硝基苯甲酸反应法:将苯乙酮与邻硝基苯甲酸在硫酸催化下反应,生成对硝基苯甲酸苯乙酮酯,再通过还原反应得到目标产物。
该方法反应步骤较多,但反应效率高,适用于大规模生产。
3. 苯乙酮与邻羟基苯甲酸反应法:将苯乙酮与邻羟基苯甲酸在酸性条件下反应,生成对羟基苯甲酸苯乙酮酯,再通过酸解反应得到2,6-二羟基苯乙酮。
该方法反应条件较为温和,适用于中小规模生产。
以上三种方法中,第一种方法是最常用的制备工艺,其产率较高且操作简单,适合工业化生产。
2,6-二羟基苯乙酮具有广泛的应用领域。
在医药领域,它是合成多种抗癌药物和抗病毒药物的重要中间体,如抗癌药物依托泊苷、阿昔洛韦等。
在染料领域,它可用于合成各种有机染料,如制备光致变色染料。
在香料领域,它可以用作合成各种香精和香料成分,如
合成芳香醇类香精。
2,6-二羟基苯乙酮是一种重要的有机合成中间体,其制备方法多样化,常用的是苯乙酮与邻氯苯甲酸反应法。
该化合物在医药、染料、香料等领域有广泛的应用,对于推动相关行业的发展和提高产品质量具有重要意义。

间硝基苯胺实验报告
实验报告
实验名称:间硝基苯胺的合成
实验地点:实验室
实验日期:2021年5月12日
实验目的:合成间硝基苯胺,了解芳香族硝化反应
实验步骤:
1.将50ml浓硫酸加入冷却至0℃的250ml棱形瓶中。
2.准确称取10g苯胺,慢慢加入硫酸中,并用磁力加热子摇床加热,使反应温度保持在0℃-5℃。
3.反应1h后,加入20ml浓硝酸,搅拌2h。
4.将反应液倒入100ml蒸馏水中,并大量加入冰块降温,收集沉淀。
5.用冰水洗涤沉淀3次,最后用冰醋酸洗涤1次,得到中间产物——间硝基偏苯胺。
6.将间硝基偏苯胺加入50ml热水中,加入10ml浓盐酸,用水浴加热至80℃,搅拌3h。
7.将得到的间硝基苯胺用水洗净,干燥得到产物。
实验结果:
1.苯胺加入硫酸中后,溶液迅速变深,放热,反应温度升高。
2.加入浓硝酸后,产生棕红色气体,反应液颜色加深。
3.洗涤后,得到沉淀物为稍带黄色的粉末状,产率为90%。
4.得到间硝基苯胺后,将其加入盐酸中,反应是强酸强碱反应,产生大量热量,溶液温度升高。
实验结论:
通过芳香族硝化反应合成了间硝基苯胺,产率为90%。
在合成
过程中,反应温度和反应物加入速度的控制非常重要,反应的过
程需要慢慢进行,并控制反应条件,才能得到较理想的产物。
在
实验中,苯胺的加入速度需要慢慢控制,反应温度需要保持在
0℃-5℃之间,加入浓硝酸时需要搅拌2h,洗涤沉淀需要注意去除
杂质。
实验中还通过强酸强碱反应,制得了较纯的间硝基苯胺。
肟菌酯中间体间三氟甲基苯乙酮的合成工艺研究偶合反应1.2 间三氟甲基苯乙酮 (3-Trifluoromethylacetophe- 四口瓶内加入乙酸乙醛肟水溶 8.0g ,50% 简称淡黄色液体主要用于医药 none)TFAP,,、农药和染料等领域它是合成甲氧基丙烯酸酯类。
液及甲苯搅拌并冷却冷却到 50.0g 100 mL ,。
5?化合物以下后开始滴加上述重氮盐清液并同时滴加,10%肟菌酯商品名的关键中间(Trifloxystrobin,Flint)硫酸铜水溶液液碱控另外通过滴加 46g,30%,体同时也是合成医药染料的重要原料国内制反应体系在通过冰盐浴控制反应,、。
pH 4~4.5,外报道的合成间三氟甲基苯乙酮的方法有间三 :温度在加完后升温至并搅拌0?~ 5?。
20?,GC 氟甲基苯甲氰与碘甲烷的格氏反应后再水解间 ;跟踪分析反应完毕后静置分层上层有机相用,,5%氨水洗至中性三氟甲基苯甲酸经酰氯化后和碘甲烷的镉试剂反。
应间三氟甲基苯甲醛和重氮甲烷反应间三氟;;水解反应1.3甲基溴苯的格氏试剂与乙酐反应间三氟甲基苯;将浓度为盐酸加入到上述有机相中90 mL 20%,加热搅拌回流跟踪分析反应水解反应完毕后胺重氮化后与乙醛肟偶合生成间三氟甲基苯乙酮,GC ,肟最后在盐酸中水解其中最后一种方法所用静置分层上层有机相用碳酸氢钠水溶液碱洗至中,。
,原料来源广涉及到的反应温度适中反应都在性无水硫酸钠干燥脱溶后再减压蒸馏得淡黄色、,,,常压下进行所有的反应都安全可控因此该方、,液体分析含量即为间三氟甲基苯乙酮47.8g,,GC ,法较易工业化其合成工艺路线如图所示笔,1 。
收率 99%,89%。
者对最后一种方法进行了相关文献的查阅并对乙醛肟再生,1.4该方法如何进一步提高收率降低成本和减少污上步水解反应的水相搅拌下加入的、27g40% 染排放进行了研究对其工业化具有一定的指导,溶液调节值至然后冷却至 NaOH pH 6.5, 10?,意义开始滴加乙醛水溶液过程中控制温。
间硝基苯甲醛的生产工艺
间硝基苯甲醛的生产工艺步骤如下:
1. 原料准备:选用丙酮、苯酚和硝酸作为主要原料。
2. 合成反应:将苯酚和丙酮在碱性条件下进行缩合反应,生成苯甲醛。
然后将苯甲醛和硝酸在硫酸催化下进行硝化反应,得到间硝基苯甲醛。
3. 精制和分离:将反应混合物进行蒸馏,将产物分离出来。
然后进行重结晶、过滤、洗涤和干燥等精制工序,获得高纯度的间硝基苯甲醛。
4. 包装贮存:将分离得到的间硝基苯甲醛进行包装,储存于干燥、阴凉、通风的地方。
上述是简略的间硝基苯甲醛的生产工艺步骤,具体情况还需根据生产企业的条件和要求等进行调整和改变。
对硝基-α-溴代苯乙酮的车间生产工艺规程目录1.产品概述 (4)1.1 化学名称 (4)1.2 产品化学结构 (4)1.3 质量标准及检验方法 (4)1.4 临床用途:有机合成中间体,是制造合霉素和氯霉素等医药的原料 (4)1.5包装规格要求及贮藏 (4)2. 设计原理 (4)2.1工艺路线选择 (4)2.2设备选型和材质选用 (5)2.3设计范围 (5)3.反应过程 (5)3.1反应机理 (5)4.生产方法及工艺规程 (5)4.1 生产 (5)4.1.1 设计采用原料 (5)4.1.2 生产工艺路线 (5)4.2化学反应式 (6)5.生产工艺工程 (6)5.1 原料配比 (6)5.2 主要工艺条件及详细操作过程 (6)5.3工艺过程简图 (6)5.4 异常现象的处理和有关注意事项 (7)5.5重点工艺控制点 (7)6 中间体和成品的质量标准和检查方法 (7)6.1 生产中间体控制项目 (7)6.2 成品出厂质量标准 (8)7.主要设备选择 (8)7.1 设备选型及选材 (8)7.1.1反应器的选择 (8)7.1.2 塔设备的选择 (9)7.1.3 换热器的选择 (9)8.生产分析 (10)8.1 说明 (10)现行的药品生产管理规范为卫生部颁布的《药品生产和质量管理规范》(GMP)8.2 车间分析任务 (11)8.2.1 原材料抽样分析 (11)8.2.2 生产中间体、半成品的分析控制 (13)8.2.3 其它分析项目 (15)9. 环境保护 (16)9.1车间环境概况 (16)9.2 车间三废处理情况 (16)10.生产安全与劳动保护 (16)10.1 有毒害物的防范措施 (16)10.2 火灾、爆炸防范措施 (17)10.3 化学灼伤害措施 (17)10.4 人身防护措施各装置 (17)11.设备一览表及主要设备运行功能 (17)12.操作工时与生产周期 (18)13.劳动组织与岗位定员 (18)14.物料平衡表 (18)15.附录 (19)1.产品概述1.1 化学名称对硝基-α-溴代苯乙酮英文名称:2-Bromo-4'-nitroacetophenone1.2 产品化学结构化学结构式:1.3 质量标准及检验方法质量标准:熔点:77℃以上水份:≤0.2%含酸:≤0.2%色泽:以实样为准分优级、合格1.4 临床用途:有机合成中间体,是制造合霉素和氯霉素等医药的原料1.5包装规格要求及贮藏包装:每包净重50kg,成品装在聚乙烯塑料袋(规格66×100)中扎口,然后分装入编织袋中扎口,挂好批号标签出厂。
Organic Syntheses, Coll. Vol. 2, p.434 (1943); Vol. 10, p.74 (1930). m-NITROACETOPHENONE
[Acetophenone, m-nitro-]
Submitted by B. B. Corson and R. K. Hazen. Checked by Roger Adams and W. W. Moyer.
1. Procedure In a 1-l. wide-mouthed Erlenmeyer flask, immersed in an ice-salt bath contained in a 2-gal. earthenware crock, is placed 150 cc. of concentrated sulfuric acid. The flask is equipped with an efficient mechanical stirrer, a small dropping funnel, and a thermometer reaching almost to the bottom of the flask (Note 1). The stirrer is started, and, when the sulfuric acid has cooled to ice temperature or below, 60 g. (0.5 mole) of pure acetophenone is slowly dropped in from the dropping funnel at such a rate (about ten minutes for the addition) that the temperature does not rise above 5° (Note 2). After the reaction mixture has cooled, this time to about −7°, the cooled (15–20°) nitrating mixture, consisting of 40 cc. (0.65 mole) of nitric acid (sp. gr. 1.42 at 15.5°) (Note 3) and 60 cc. of concentrated sulfuric acid, is added through the dropping funnel at such a rate (100–120 drops per minute) that the temperature of the reaction mixture remains at 0° or lower (Note 4). After the nitrating acid has been added, stirring is continued for ten minutes longer and the contents of the flask are poured (Note 5), with vigorous manual stirring, into a mixture of 750 g. of cracked ice and 1.5 l. of water. The product separates as a yellow flocculent solid.
After the ice has melted, the product is filtered by suction and the somewhat sticky mass pressed as dry as possible. It is transferred to a mortar and triturated with three successive 300-cc. portions of cold water (to remove acid); it is then stirred to a mush with two successive 25-cc. portions of ice-cold ethyl alcohol (to remove adhering oil); the solid is pressed dry on the suction filter after each of the five washings. The product is pressed on a porous plate and, when fairly dry (about one hour), is dissolved in 100–120 cc. of hot ethyl alcohol (Note 6). The dark solution is filtered quickly through a small suction funnel (Note 7), and the hot filtrate is poured slowly into 1 l. of cold water which is stirred vigorously (Note 8) with a stirring rod during the addition and for several minutes afterward. After standing a few minutes the yellow solid is filtered by suction, washed with 200 cc. of cold water, sucked dry, and pressed out on a porous clay plate.
When the reprecipitated m-nitroacetophenone (50–55 g.) is dry it is dissolved in 100 cc. of hot alcohol in a 300-cc. Erlenmeyer flask. The flask is then immersed in an ice bath and shaken vigorously while crystallization is taking place. Because of the great change in solubility between 60° and 50° the agitation of the liquid must be vigorous during this temperature interval, or large clumps of crystals will be formed instead of the purer and more easily dried mush. After the temperature has reached 20° (about one hour) the mixture is filtered by suction (Note 9). The solid (Note 10) is washed with 10 cc. of ice-cold alcohol and pressed dry on a clay plate. The product is light yellow; it softens at 74° and melts at 76–78°. The total yield is 45 g. (55 per cent of the theoretical amount) (Note 11).
2. Notes 1. The only flask found suitable for this preparation is the 1-l. wide-mouthed Erlenmeyer. A large flask is necessary to ensure rapid cooling. A propeller with long, wide bladesagitates the viscous liquid much more efficiently than a stirrer of the centrifugal type. The blades should be as long as allowed by the wide mouth of the flask. The thermometer which is to record the temperature of the reaction mixture should enter at an angle and reach almost to the bottom of the flask, since the amount of liquid is small. The thermometer should have the zero point at least 15 cm. from the bulb in order to facilitate reading the temperature. It is essential that the temperature be watched throughout the experiment. A smaller ice-salt bath than that contained in a 2-gal. earthenware crock is inadequate. 2. The same dropping funnel can be used, without washing, for the addition of the concentrated sulfuric acid, the acetophenone, and the nitrating mixture. With rapid stirring the acetophenone can be easily added in seven minutes without raising the temperature of the reaction mixture above 3°. 3. Nitric acid of lower specific gravity than 1.42 at 15.5° yields an impure product. Ordinary concentrated nitric acid usually has to be strengthened by the addition of fuming nitric acid. 4. Two conditions are necessary for a good nitration: low temperature (0° or below) and rapidity of addition of the nitrating mixture (not longer than forty-five minutes). With efficient cooling the temperature can be held between −5 and 0°. If the temperature should rise once or twice to 3° no harm is done provided that the reaction mixture is quickly cooled back to the correct temperature. In order to avoid local heating the delivery tube of the dropping funnel should be so directed as to deliver the nitrating acid near the site of greatest agitation. The rate of stirring must be rapid. The optimum speed will be different for different stirrers, depending on the shape and size of the blades. For the stirrer described the speed is 1600 r.p.m. During the addition of acetophenone this high speed is not necessary; in fact, it cannot be maintained on account of excessive splashing. However, during the addition of the nitrating acid the reaction mixture thickens and high-speed stirring becomes possible. The ice-salt mixture must be stirred repeatedly with a stick, and fresh ice and salt must be added from time to time. The temperature of the bath should be around −16°. Enough liquid should be present in the cooling bath so that the lower half of the flask is immersed in brine. If no especial care is exercised, the addition of the nitrating acid requires from two to three hours in order to maintain the temperature at 0° or below. This long exposure to the mixture of sulfuric and nitric acids is as harmful as a rise of temperature; the product is of poor quality and the yield drops from 55 to 15 per cent or less. By using solid carbon dioxide as the cooling medium and also adding it directly to the reaction mixture, a temperature of −20° can be maintained during the nitration.1 According to W. W. Hartman, private