蛋白质结构域数据库
- 格式:ppt
- 大小:3.40 MB
- 文档页数:74


生物信息研究中常用蛋白质数据库简述内蒙古工业大学理学院呼和浩特孙利霞2010.1.5摘要:在后基因组时代生物信息学的研究当中,离不开各种生物信息学数据库。
尤其在蛋白质从序列到功能的研究当中,目前各种行之有效的方法都是基于各种层次和结构的蛋白质数据库。
随着计算机技术及网络技术的发展,目前的蛋白质数据库不论是所包含数据量还是功能都日新月异,新的数据库层出不穷。
一个新手面对如此浩瀚的数据量往往无从下手。
本文粗浅地为目前蛋白质数据库的使用勾画出一个轮廓,作为自己蛋白质研究入门的一个引导。
关键词:蛋白质;数据库0 引言随着科技的发展,个人的知识往往赶不上快速膨胀的信息量,人们为了解决这个问题,便创建了形形色色的数据库。
蛋白质数据库是指:在蛋白质研究领域根据实际需要,对蛋白质序列、蛋白质结构以及文献等数据进行分析、整理、归纳、注释,构建出具有特殊生物学意义和专门用途的数据库。
蛋白质数据库总体上可分为两大类:蛋白质序列数据库和蛋白质结构数据库,蛋白质序列数据库来自序列测定,结构数据库来自X-衍射和核磁共振结构测定(详见图1)。
这些数据库是分子生物信息学的基本数据资源。
上世纪90年代,我国从事蛋白质研究的学者使用的蛋白质数据库储存介质还是国外实验室发布的激光光盘[1]。
信息的传播储存甚为不便。
随着蛋白质研究的发展飞快,同时伴随着计算机和因特网发展,蛋白质数据库的储存传播方式也发生的巨大的变化。
进入21世纪后,我们所用的各种蛋白质数据库都发展成为存储在网络服务器上,基于“服务器—客户机”的访问查询方式。
伴随着计算机及物理测试技术的发展数据库的容量和功能成数量级膨胀。
但是面对如此浩瀚的数据,新手往往感到无从下手,在需要时找不到自己需要的合适数据库。
本文从目前蛋白质数据库建立的的逻辑层次出发,系统地简绍了常用蛋白质数据的概况,它们的查询方法以及它们相互之间的联系。
同时尽量不涉及数据库建设和维护方面的计算机和网络这些数据库底层的技术,为蛋白质研究的入门者及对蛋白质感兴趣的人员的一个引导。


蛋白质结构域名词解释
蛋白质结构域是指蛋白质分子中具有特定结构和功能的独立模块。
这些结构域通常由100~300个氨基酸残基组成,可以折叠成稳定的三
维结构,并参与蛋白质的特定功能,如结合其他分子、催化化学反应、信号传导等。
蛋白质结构域的存在使得蛋白质能够在不同的生物过程
中发挥各种不同的功能。
蛋白质结构域的发现对于我们理解蛋白质的功能和进化具有重要
意义。
通过研究蛋白质结构域,科学家们可以预测蛋白质的功能、相
互作用和信号传导途径。
此外,蛋白质结构域的模块化性质使得研究
人员能够重新组合和改变蛋白质的功能,从而设计出具有特定功能的
蛋白质。
蛋白质结构域可在蛋白质数据库中进行存储和查询。
目前已经发
现了许多不同的蛋白质结构域,其中一些具有广泛的功能和分布,而
另一些则是特定物种或特定蛋白质家族所特有的。
研究人员可以通过
比对已知的蛋白质结构域来预测新发现的蛋白质中是否存在类似的结
构域,并进一步探究其功能和作用机制。
总之,蛋白质结构域是蛋白质分子中具有特定结构和功能的独立模块,对于研究蛋白质功能和设计功能性蛋白质具有重要意义。
通过研究蛋白质结构域,我们能够深入了解蛋白质的功能和相互作用,为生物学、药物研发等领域的研究提供了基础。
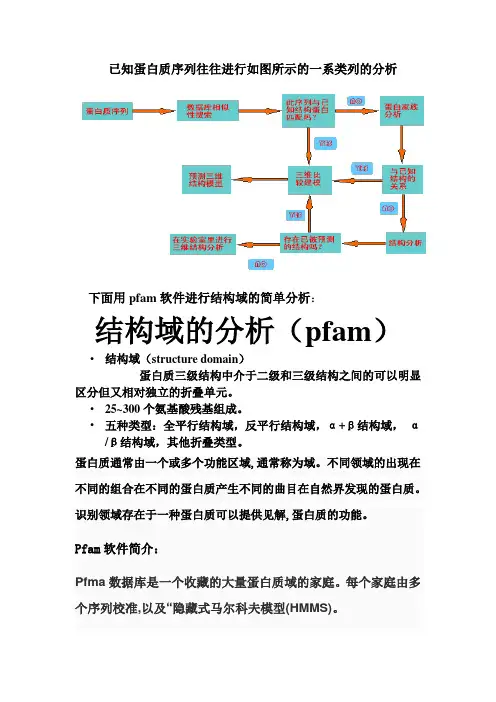
已知蛋白质序列往往进行如图所示的一系类列的分析下面用pfam软件进行结构域的简单分析:结构域的分析(pfam)•结构域(structure domain)蛋白质三级结构中介于二级和三级结构之间的可以明显区分但又相对独立的折叠单元。
•25~300个氨基酸残基组成。
•五种类型:全平行结构域,反平行结构域,α+β结构域,α/β结构域,其他折叠类型。
蛋白质通常由一个或多个功能区域,通常称为域。
不同领域的出现在不同的组合在不同的蛋白质产生不同的曲目在自然界发现的蛋白质。
识别领域存在于一种蛋白质可以提供见解,蛋白质的功能。
Pfam软件简介:Pfma数据库是一个收藏的大量蛋白质域的家庭。
每个家庭由多个序列校准,以及“隐藏式马尔科夫模型(HMMS)。
有两个等级的品质,pfam家庭:一个和Pfma b。
Pfma a条目包含了许多来自底层序列数据库,称为Pfam seq,这是由最近发布的UniProtKB在给定的时间点。
Pfam a家庭由许多一个策划种子含有少量的对齐代表家族成员,剖面隐马尔可夫模型(HMMS)由种子对齐和一个自动生成的全排列,其中包含所有蛋白质序列检测属于家庭定义为HMM搜索数据库的主序列。
Pfam b家庭联合国注释和低质量为他们从集群自动生成非冗余的最新加入释放。
尽管低的质量,pfam b家庭可以用于识别功能守恒的地区没有发现了pfam a一个条目。
•利用pafm进行蛋白质结构域及功能位点分析MSRQAWIETSALIECISEYGTKCSFDTFQGLTINDISTLSNLM NQISV ASVGFLNDPRTPLQAMSCEFVNFISTADRHAYMLQK NWFDSDV APNVTTDNFIATYIKPRFSRTVSDVLRQVNNFALQ PMENPKLISRQLGVLKAYDIPYSTPINPMDV ARSSANVVGNV SQRRALSTPLIQGAQNVTFIVSESDKIIFGTRSLNPIAPGNFQI NVPPWYSDLNVVDARIYFTNSFLGCTIQNVQVNA VNGNDPV ATITVPTDNNPFIVDSDSVVSLSLSGGAINVTTA VNLTGYAIAI EGKFNMQMNASPSYYTLSSLTIQTSVIDDFGLSAFLEPFRIR LRASGQTEIFSQSMNTLTENLIRQYMPANQA VNIAFVSPWY RFSERARTILTFNQPLLPFASRKLIIRHLWVIMSFIA VFGRYY TVNkeywor d seaSignificant Pfam-A Matches Show or hide all alignments.Description EntrytypeClanEnvelope AlignmentStart End Start Endefhand_like Phosphoinositide-specificphospholipase C, efhand-likeDomain C L0220 245 318 250 318PI-PLC-X Phosphatidylinositol-specificphospholipase C, X domainFamily CL0384 322 465 322 465SH2SH2 domain Domain n/a550 639 550 639 SH2SH2 domain Domain n/a668 741 668 741 SH3_1SH3 domain Domain C L0010 797 843 797 843PI-PLC-Y Phosphatidylinositol-specificphospholipase C, Y domainFamily CL0384 952 1070 952 1070C2C2 domain Domain C L01541090 1177 1092 1176 nsignificant Pfam-A Matches Show or hide all alignments.Family Description EntrytypeClanEnvelope Alignment HMMBiscoStart End Start End From ToPH PH domain Domain C L026634 142 40 139 8 101 24.nsignificant Pfam-A Matches Show or hide all alignments.EF_hand_4EF-handdomainDomain C L0220 156 192 157 183 2 28 10.PH PH domain Domain C L0266 489 575 490 533 2 38 13.PH PH domain Domain C L0266 842 931 873 929 44 102 16. Pfam-B Matches Show or hide all alignments.Pfam-B_12554n/a n/a n/a37 232 45 164#HMM kPKfcpfrLssDesaLiWyskkkeKr..lkLSsvsriiiGqrTavFery....lrpeke #MATCH +P f++ +++++W + + + + + + +i +G+ + F ry + + #PP 5777788888888*****9996555566*********************5443223468 #SEQ R PERKTFQVKLETRQITWSRG ADKIEga IDIREIKEIRPGKTSRDFDRY qedpAFRPD QComments or questions on the site? Send a mail to pfam-help@。



TBtools蛋白结构域1. 简介TBtools(Tools for Biologists)是一个用于生物学数据分析和可视化的集成工具。
其中,TBtools蛋白结构域模块提供了一系列功能,用于分析和可视化蛋白质结构域。
2. 蛋白结构域的概念蛋白质结构域是指具有一定功能和稳定空间结构的蛋白质片段。
它们通常由连续的氨基酸序列组成,并且在进化过程中被保留下来。
蛋白质结构域可以通过许多不同的方法进行识别和分类,例如序列比对、结构预测和功能注释等。
3. TBtools蛋白结构域功能3.1 蛋白结构域识别TBtools提供了多种常用的蛋白质结构域识别工具,例如HMMER、InterProScan、SMART等。
用户可以根据需要选择适当的工具进行分析。
这些工具可以基于不同的算法和数据库对输入的蛋白质序列进行扫描,并输出相应的结构域信息。
3.2 结果可视化TBtools提供了多种可视化方式,帮助用户更直观地理解和分析蛋白质结构域。
其中包括:•蛋白质结构域图:将蛋白质序列上的结构域标注在一条线上,颜色和形状表示不同的结构域类型。
用户可以通过缩放、平移和旋转等操作来查看详细信息。
•蛋白质结构域热图:将多个蛋白质序列的结构域信息绘制成热图,颜色表示结构域的存在与否以及其相对位置。
这种可视化方式可以帮助用户比较不同蛋白质之间的结构域差异。
•蛋白质结构域树:将蛋白质序列根据其结构域组成进行聚类,并绘制成树状图。
这种可视化方式可以帮助用户发现具有相似结构域组成的蛋白质群体。
3.3 结果分析TBtools还提供了一些功能,用于进一步分析和挖掘蛋白质结构域的信息。
其中包括:•结构域注释统计:统计输入序列中各个结构域类型的数量和频率,并生成统计图表。
用户可以通过这些统计信息来了解不同结构域类型的分布情况。
•结构域功能富集分析:根据结构域注释和GO、KEGG等数据库,对结构域的功能进行富集分析。
富集分析可以帮助用户发现与特定生物学过程或通路相关的结构域。

填空题:1、蛋白质结构数据来源:①实验测定方法: X-ray 、 NMR 、Cryo-EM ②理论预测:同源建模、折叠识别、从头计算2、一级数据库:①一级核酸数据库:Genbank(美国)、EMBL (欧洲)、DDBJ(日本) NCBI②一级蛋白质序列数据库:SWISS-PORT 、PIR 、 NCBI③一级蛋白质结构数据库:PDB、 pfam 、 prosite大分子序列格式:fasta数据库基本文件格式:genbank蛋白质分类数据库:SCOP、CATH 、 FSSP二次数据库: GDB 、 Prosite、 TRANSFAC3、本地软件: Clustal-x 、 BioEdit 、 Mega、 sequencher、 spdbv、 Discovery-studio4、本课程主要理论依据:相似性、同源性、序列比对(3D结构比对)、数学方法、分子动力、分子力学5、基因鉴定三步骤:①找到序列中的非编码区(低复杂度区)②找基因③鉴定找到的基因6、主要的生物大分子数据:①DNA:基因组序列、基因序列、cDNA、EST、碱基修饰DNA 功能模块 /位点(如启动子、剪接体、表达调控位点等)②蛋白质:氨基酸组成、氨基酸序列、理化性质、原子坐标;二级结构、核体、结构域、功能域 /位点; 3D 结构常见的生物信息数据记录格式:FASTA 、GenBank、EMBL、 PDBFASTA 格式:序列文件的第一行由大于符号>大头的任意文字说明,主要为标记序列用。
从第二行开始是序列本身,标准核苷酸符号或氨基酸单字母符号,通过核苷酸符号大小写均可,而氨基酸一般用大写字母。
文件中和每一行都不要超过80 个字符(通常60 个字符)GenBank格式:序列名称、长度。
日期;序列说明、编号、版本号;物种来源、学名、分类60学位置;相关文献作者、题目、刊物、日期;序列特征表;碱基组成;序列本身(每行个)二 .填空题1.常用的三种序列格式: NBRF/PIR,FASTA 和 GDE2.初级序列数据库: GenBank, EMBL 和 DDBJ3.蛋白质序列数据库: SWISS-PROT 和 TrEMBLPIR (蛋白4. 提供蛋白质功能注释信息的数据库:KEGG (京都基因和基因组百科全书)和质信息资源) 5. 目前由 NCBI 维护的大型文献资源是PubMed6.数据库常用的数据检索工具: Entrez, SRS, DBGET7.常用的序列搜索方法: FASTA 和 BLAST8.高分值局部联配的 BLAST 参数是 HSPs(高分值片段对), E(期望值) 9. 多序列联配的常用软件: Clustal10.蛋白质结构域家族的数据库有:Pfam, SMART11. 系统发育学的研究方法有:表现型分类法,遗传分类法和进化分类法12. 系统发育树的构建方法:距离矩阵法,最大简约法和最大似然法13. 常用系统发育分析软件:PHYLIP 14.检测系统发育树可靠性的技术: bootstrapping 和 Jack-knifing 15. 原核生物和真核生物基因组中的注释所涉及的问题是不同的16. 检测原核生物ORF 的程序: NCBI ORF finder17. 测试基因预测程序正确预测基因的能力的项目是GASP(基因预测评估项目)18.二级结构的三种状态:α螺旋,β折叠和β转角19.用于蛋白质二级结构预测的基本神经网络模型为三层的前馈网络,包括输入层,隐含层和输出层20.通过比较建模预测蛋白质结构的软件有SWISS-PDBVIEWER ( SWISS — MODEL 网站) 21. 蛋白质质谱数据搜索工具:SEQUEST 22. 分子途径最广泛数据库:KEGG23. 聚类分析方法,分为有监督学习方法,无监督学习方法24. 质谱的两个数据库搜索工具:1、 SEQEST 和 Lutkefi 三大数据库:核酸序列数据库、蛋白质序列数据库、结构数据库世界三大核酸序列数据库:GenBank、 EMBL-Bank 、 DDBJ蛋白质序列数据库:Swiss-Prot、 TrEMBL 、UniProt蛋白质结构数据库:PDB 、SCOP、CATH2、 GenBank 文献、提供了提供的服务:提供了EntrezBLAST 序列类似性检索。

蛋白结构域互作分析方法蛋白结构域互作分析是研究蛋白质相互作用的重要方法之一、蛋白结构域是蛋白质分子中的一段连续的高保守序列,具有特定的结构和功能。
通过分析蛋白质结构域之间的相互作用,可以揭示蛋白质互作网络的拓扑结构,进而理解蛋白质功能及其在细胞内信号传导、转录调控、蛋白质合成、代谢调控等生物过程中的作用。
本文将介绍几种常用的蛋白结构域互作分析方法。
1. 结构域相互作用数据库的分析方法:结构域相互作用数据库存储了已知的蛋白质结构域组合的信息,如DIP、BIND、IntAct等。
通过结构域相互作用数据库,可以获取蛋白结构域之间的已知相互作用信息,进而预测相关蛋白之间的互作关系。
这种方法主要依赖于结构域相互作用数据库的积累和维护,具有较高的可靠性。
2.蛋白互作预测方法:蛋白互作预测方法通过分析蛋白序列中的保守结构域进行预测。
保守结构域是指在进化过程中高度保守的结构域,其具有相似的结构和功能。
在预测蛋白互作时,首先利用多序列比对和引擎等工具识别出蛋白序列中的保守结构域,然后通过比较已知互作蛋白结构域与待预测蛋白结构域之间的相似性,来预测它们之间的互作潜力。
4.结合实验方法的结构域互作分析:结合实验方法的结构域互作分析是通过实验手段来验证已预测或研究的蛋白结构域之间的相互作用关系。
常见的实验方法包括酵母双杂交、共沉淀、共免疫沉淀、亲和层析等。
通过这些实验方法,可以验证预测蛋白结构域的相互作用关系,并获取更全面、可靠的互作信息。
综上所述,蛋白结构域互作分析方法包括结构域相互作用数据库的分析方法、蛋白互作预测方法、结构域互作网络分析方法以及结合实验方法的结构域互作分析。
这些方法通过不同的途径揭示了蛋白质结构域之间的相互作用网络,为深入理解蛋白质功能及其在生物过程中的作用提供了重要的分析工具。
蛋白质结构分析原理及工具(南京农业大学生命科学学院生命基地111班)摘要:本文主要从相似性检测、一级结构、二级结构、三维结构、跨膜域等方面从原理到方法再到工具,系统地介绍了蛋白质结构分析的常用方法。
文章侧重于工具的列举,并没有对原理和方法做详细的介绍。
文章还列举了蛋白质分析中常用的数据库。
关键词:蛋白质;结构预测;跨膜域;保守结构域1 蛋白质相似性检测蛋白质数据库。
由一个物种分化而来的不同序列倾向于有相似的结构和功能。
物种分化后形成的同源序列称直系同源,它们通常具有相似的功能;由基因复制而来的序列称为旁系同源,它们通常有不同的功能[1]。
因此,推测全新蛋白质功能的第一步是将它的序列与进化上相关的已知结构和功能的蛋白质序列比较。
表一列出了常用的蛋白质序列数据库和它们的特点。
表一常用蛋白质数据库网址可能有更新氨基酸替代模型。
进化过程中,一种氨基酸残基会有向另一种氨基酸残基变化的倾向。
氨基酸替代模型可用来估计氨基酸替换的速率。
目前常用的替代模型有Point Accepted Mutation (PAM)矩阵、BLOck SUbstitution Matrix (BLOSUM)矩阵[2]、JTT模型[3]。
序列相似性搜索工具。
序列相似性搜索又分为成对序列相似性搜索和多序列相似性搜索。
成对序列相似性搜索通过搜索序列数据库从而找到与查询序列相似的序列。
分为局部联配和全局联配。
常用的局部联配工具有BLAST和SSEARCH,它们使用了Smith-Waterman 算法。
全局联配工具有FASTA和GGSEARCH,基于Needleman-Wunsch算法。
多序列相似性搜索常用于构建系统发育树,这里不阐述。
表二列举了常用的成对序列相似性比对搜索工具表二成对序列相似性比对搜索工具网址可能有更新2 蛋白质一级结构分析(含保守结构域)蛋白质结构的基本信息来源于它的一级结构,分析蛋白质一级结构的第一步是将它们分成其组成部分,然后处理每个部分的结构[4]。
⽣物数据库介绍——NCBINCBI(National Center for Biotechnology Information,美国国家⽣物技术信息中⼼)除了维护GenBank核酸序列数据库外,还提供数据分析和检索资源。
NCBI资源包括Entrez、Entrez编程组件、MyNCBI、PubMed、PudMed Central、PubReader、Gene、the NCBI Taxonomy Browser、BLAST、Pimer-Blast、COBALT、RefSeq、UniGene、HomoloGene、ProtEST、dbMHC、dbSNP、dbVar、Epigenomics、the Genetic Testing Registry、Genome和相关⼯具、⽐对查看器、跟踪存档、Sequence Read Archive、BioProject、BioSample、ClinVar、MedGen、HIV-1/⼈类蛋⽩质相互作⽤数据库、Gene Expression Omnibus、Probe、Online Mendelian Inheritance in Animals、the Molecular Modeling Database、the Conserved Domain Database、the Conserved Domain Architecture Retrieval Tool、Biosystem、Protein Clusters and thePubChem suite of small molecule databases,所有这些资源可以在NCBI主页找到。
Databases⼀个提供有关基因组组装结构,装配名称和其他元数据,统计报告以及基因组序列数据链接等信息的数据库。
⼀个有关培养物、动植物样本和其他⾃然样本的精选元数据集。
记录显⽰样本状态,有关馆藏的机构的信息,以及NCBI中相关数据链接。
蛋白质序列、性质、功能和结构分析基于网络的蛋白质序列检索与核酸类似,从NCBI或利用SRS系统从EMBL检索。
1、疏水性分析ExPASy的ProtScale程序(/cgi-bin/protscale.pl)可用来计算蛋白质的疏水性图谱。
输入的数据可为蛋白质序列或SWISS-PROT数据库的序列接受号。
也可用BioEdit、DNAMAN等软件进行分析。
2、跨膜区分析蛋白质跨膜区域分析的网络资源有: TMPRED:/software/TMPRED_form.html PHDhtm: http:www.embl-heidelberg.de/Services/ ... predictprotein.html MEMSAT: ftp://3、前导肽和蛋白质定位一般认为,蛋白质定位的信息存在于该蛋白自身结构中,并且通过与膜上特殊受体的相互作用得以表达。
这就是信号肽假说的基础。
这一假说认为,穿膜蛋白质是由 mRNA编码的。
在起始密码子后,有一段疏水性氨基酸序列的RNA片段,这个氨基酸序列就称为信号序列(signal sequence)。
蛋白质序列的信号肽分析可联网到http://genome.cbs.dtu.dk/services/SignalP/或其二版网址http: //genome.cbs.dtu.dk/services/SignalP-2.0/。
该服务器也提供利用e-mail 进行批量蛋白质序列信号肽分析的方案(http://genome.cbs.dtu.dk/services/SignalP/mailserver.html),e-mail 地址为 signalp@ genome.cbs.dtu.dk。
蛋白质序列中含有的信号肽序列将有助于它们向细胞内特定区域的移动,如前导肽和面向特定细胞器的靶向肽。
在线粒体蛋白质的跨膜运输过程中,通过线粒体膜的蛋白质在转运之前大多数以前体形式存在,它由成熟蛋白质和N端延伸出的一段前导肽或引肽(leader peptide)共同组成。
分子生物学中常用数据库综合数据库:来源:/news/science/article/90048.html生物信息学网址链接:http://www.bioinformatics.ca/links_directory/Nucleic Acid Research Database Issue:/content/vol32/suppl_2/一、蛋白相关数据库蛋白质结构域预测工具Esignal:/esignal/信号传导系统蛋白的结构域预测工具,凡是涉及到信号传导系统的蛋白用这个预测效果最佳SignalP:http://www.cbs.dtu.dk/services/SignalP/信号肽预测工具,适合定位于非胞质位置的蛋白质Emotif:/emotif-search/结构域预测工具,由于其用motif电子学习的方法产生结构域模型,故预测效果比Prosite好Ematrix:/ematrix/是用Matrix的方法创建的结构域数据库,可与emotif互相印证。
其速度快,可快速搜索整个基因组InterPro:/InterProScan/EBI提供的服务,用图形的形式表示出搜索的结构域结果TRRD:http://wwwmgs.bionet.nsc.ru/mgs/gnw/trrd/转录因子结构域预测的最好数据库。
但不会用Protscale:/cgi-bin/protscale.pl可分析该序列的各种性状如活动度、亲水性(Kyte&Doolittle)、抗原性(Hopp&Woods)等通过寻找MOTIF和Domain来分析蛋白质的功能A. MOTIF是蛋白中较小的保守序列片断,其概念比Domain小PROSITE:/tools/scanprosite/是专门搜索蛋白质Motif的数据库,其中signature seqs是最重要的motif信息B. Domain:若干motif可形成一个Domain,每个Domain形成一个球形结构,Domain与Domain之间通常像串珠一样相连Pfam:可以搜索某段序列中的Domain,并以图形化表示出来。