表达谱芯片数据分析项目示例
- 格式:pdf
- 大小:338.66 KB
- 文档页数:3


第18卷第6期微阵列技术[1-3]的到来对生物学和医学来说是一场革命,通过它可以同时观测成千上万个基因的表达水平,从而能够在基因组水平上以系统的、全局的观念去研究生命现象及其本质。
还可以根据基因在不同条件下表达的差异性来进行复杂疾病诊断、药物筛选、个性化治疗、基因功能发现、农作物优育和优选、环境检测和防治、食品卫生监督及司法鉴定等,因此对基因表达谱的研究具有重要的理论价值和应用意义。
微阵列基因表达数据具有维数高、样本小、非线性的特点,这对一些传统的机器学习方法提出了新的挑战,对其数据的分析已成为生物信息学研究的焦点。
1基因表达数据采集基因表达数据采集可分为三个步骤:微阵列设计、图像分析和数据获取、过滤、标准化。
基因芯片(gene chip ),简称为微阵列,就是指固着在载体上的高密度DNA 微点阵,具体地说就是将靶基因或寡核苷酸片段有序地、高密度排列在玻璃、硅等载体上。
mRNA (信使核糖核酸)的表达水平的获得是通过选取来自不同状态的样本(如正常组织与肿瘤组织、不同发育阶段组织,或用药之前与用药之后组织等,一种称为实验样本,另外一种称为参考样本),在逆转录过程中,实验样本和参考样本RNA (核糖核酸)分别用不同的红、绿荧光染料去标记,并将它们混合,与微阵列上的探针序列进行杂交,经适当的洗脱步骤与激光扫描仪对芯片进行扫描,获得对应于每种荧光的荧光强度图像,通过专用的图像分析软件,可获得微阵列上每个点的红、绿荧光强度(Cy5和Cy3),其比值(Cy5/Cy3)表示该基因在实验样本中的表达水平。
在通常情况下,考虑Cy5和Cy3的数值时,还应考虑相应的背景数值,如果微阵列上某个基因的Cy5或Cy3数值比相应的背景数值低,则该基因的表达水平无法确定。
为了方便数据处理,常孟令梅等:一种基于DCT 变换的图像认证算法文章编号:1005-1228(2010)06-0017-03基因表达谱数据分析技术刘玲(江苏财经职业技术学院,江苏淮安223001)摘要:人类基因组计划的研究已进入后基因组时代,后基因组时代研究的焦点已经从测序转向功能研究,主要采用无监督和有监督技术来分析基因表达谱和识别基因功能,通过基因转录调控网络分析细胞内基因之间的相互作用关系的整体表示,说明生命功能在基因表达层面的展现,对目前基因表达谱数据分析技术及它们的发展,进行了综述性的研究,分析了它们的优缺点,提出了解决问题的思路和方法,为基因表达谱的进一步研究提供了新的途径。

基因表达谱分析的实验方法及数据解读基因是生物体内最基本的生物学信息单元,它们的表达水平可以反映生物活动的差异性。
为了更好地了解基因表达的机制,越来越多的科学家开始关注基因表达谱分析。
通过基因表达谱分析,我们可以了解基因的表达情况以及基因与疾病相关的信息。
本文将从实验方法和数据解读两个方面进行介绍,帮助读者更好地了解基因表达谱分析。
一、实验方法1. 前期准备基因表达谱分析需要进行实验,而实验的准备工作非常重要。
首先,必须选择要研究的样本,如人类组织、小鼠细胞、大麦品种等。
因为样本数量和质量对结果的影响非常大,因此在选择样本时必须严谨。
其次,为了确保数据的准确性和可重复性,必须严格按照实验流程操作。
如RNA提取、RNA浓度、DNA酶处理等步骤,如果有一步出错,就会影响整个实验的结果。
最后,选择适当的实验方法也非常重要,可以根据研究的目的和研究条件选择不同的方法。
2. 基本实验方法(1)Microarray分析Microarray分析是一种快速高通量的DNA分析技术,它可以同时分析成千上万个基因在不同条件下的表达水平。
使用这种方法需要用特定的芯片进行实验,芯片的制作需要基因组数据和探针的设计。
该方法可以发现全局基因的表达差异,但是只能分析已知基因,因此对于基因组结构不完整的生物来说不是很适用。
(2)RNA-seq分析RNA-seq分析是一种利用高通量测序技术的快速分析RNA的方法。
使用这种方法需要进行RNA的提取、建库、测序,然后通过数据分析得到基因表达谱。
与Microarray相比,这种方法可以分析未知基因和表达水平较低的基因,因此适用于各种不同生物的表达分析。
二、数据解读1. 数据聚类和热图分析一般来说,在基因表达数据处理中,处理出来的基因表达数据大小可能会很大,观察起来非常困难,不方便数据分析和判断。
因此,聚类分析和热图是可视化数据的常用方式。
聚类可以将基因根据其表达水平分为不同的类别,所以可以更好地理解垂直方向上类别的信息。


基因表达数据分析方法及其应用研究共3篇基因表达数据分析方法及其应用研究1随着技术的不断发展,基因表达数据分析在生命科学研究中扮演着越来越重要的角色。
基因表达数据分析是研究基因功能的关键一步,它使得科学家可以了解基因在特定情况下的表达水平。
在本文中,我们将讨论基因表达数据分析的方法及其应用。
1.基因表达数据的来源和类型基因表达数据是通过分析转录组和基因芯片等数据获得的。
转录组技术通过测量RNA浓度,包括RNA-seq和microarray。
而基因芯片就是一种将成千上万的基因测量并呈现的芯片。
基因表达数据存在多种类型,包括原始数据、表达矩阵、差异表达矩阵、注释文件和元数据等等。
2. 基因表达数据分析的方法(1)数据清理数据清理是数据分析过程中的第一步。
它包括数据预处理、去除冗余数据、去除噪声和填补数据空缺等操作。
(2)正则化正则化的目的是调整不同基因表达数据之间的差异,消除数据中的计量误差和探测效率的误差。
几种正则化方法包括平滑、归一化和标准化。
(3)差异分析差异分析是研究基因表达数据中各基因在不同样品之间差异的方法。
常用的差异分析方法包括t-test、ANOVA、FDR和q值等。
(4)聚类分析聚类分析是将数据根据观察指标相似度进行分类的方法。
在基因表达数据上,它通常用于发现不同条件下的基因表达模式。
(5)变异分析变异分析是一种寻找表达值变异的基因的方法。
通常,基因的变异程度与其在癌症和其他疾病中的作用有关。
(6)功能注释功能注释是将基因表达数据与已知基因功能相结合的方法,从而获得数据更深层次的信息。
它通常用于解释基因表达数据的生物学意义,如基因表达数据和肿瘤发展的相关性等。
3.应用研究基因表达数据分析可应用于许多研究领域,包括基因表达和调控、单细胞分析和肿瘤生物学等。
(1)基因表达和调控基因表达数据分析可用于挖掘基因之间的相互关系以及调控通路。
这些信息可以在理解细胞生物学、发育及疾病发生机制的过程中发挥重要作用。

第61章 基因表达谱的概念与数据分析技术目前,大部分基于芯片的研究主要是监控基因表达水平,获得基因表达图谱。
基因表达是根据基因的DNA 模板进行mRNA 和蛋白质合成的过程。
基因芯片能够研究差异表达基因,找到基因调控网络及其机制,揭示不同层次多基因协同作用的生命过程。
表达型基因芯片将在研究人类重大疾病如癌症、心血管病等相关基因及作用机理方面发挥巨大作用。
本章主要就基因表达谱的概念和常用的数据分析技术作简要的介绍,并利用SAS 软件对实例数据进行分析。
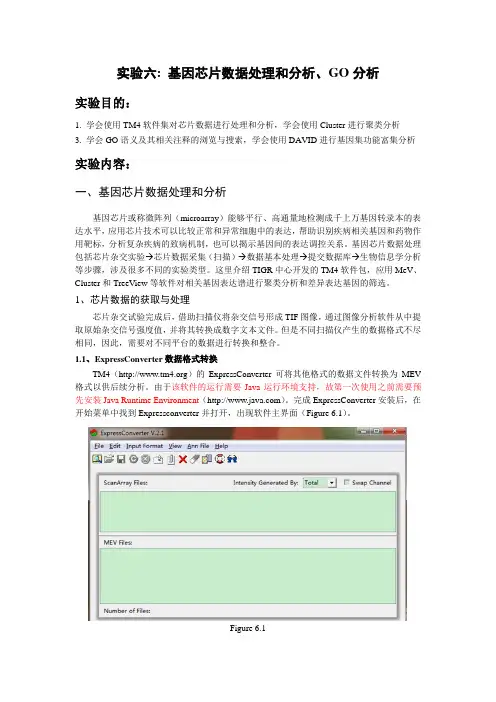
61.1基因表达谱的概念61.1.1 基因芯片基因芯片是通过反应体系中不同来源的DNA 与芯片探针的竞争性杂交获得检测信号,得到的检测数据是两个信道荧光强度的比值,如图61-1和图61-2。
一张芯片经过标准化以后,该芯片上的每一个基因都可以获得一个“表达比(ratio )”,表达比表示在某一实验条件下,某一基因在样品中的表达强度与其在对照中的表达强度的比值,通常采用表达比的对数形式,如2log ()ratio 能够更直观地描述基因表达上调或下调的幅度。
图61-1 基因芯片数据的产生流程图图61-2 基因芯片spots61.1.2 基因表达图谱与空间由若干次芯片实验可以得到一个m n ×的基因表达矩阵(gene expression matrix ),用以记录基因在不同的实验条件下的表达图谱,如图61-3。
通常m 为基因的数目,n 为实验的次数或芯片的数目。
表达矩阵也可以理解为一个表达空间,每一次实验都代表空间中一条单独的、不同的轴。
表达矩阵中每一行的数据即为该基因的表达向量,对每一个基因而言,表达向量描述了其在“表达空间”中的位置,某次实验某一个基因的2log ()ratio 值代表这个基因的几何坐标,这样一个基因就可以被描述成表达空间中的一个点。
从这个观点出发来解释基因表达,如果两个基因在每一个实验中有相近的2log ()ratio 值,那么在表达空间中两个基因的点会非常接近;相反,则两个基因的点会相距较远,如图61-4。

实验6基因芯片数据处理分析与GO分析实验背景:基因芯片技术是通过检测靶基因在不同样本中的表达量差异,并分析其生物信息学特性,来揭示基因调控网络与疾病发生发展的过程的一种高通量技术。
基因芯片数据处理和分析是基因芯片研究的关键步骤之一、通过对基因芯片数据进行预处理、差异分析、聚类分析等,可以获得与研究目标相关的基因列表,并进一步进行GO(Gene Ontology)的功能富集分析,揭示差异表达基因的功能特性。
实验目的:通过基因芯片数据处理分析和GO功能富集分析,获得与研究目标相关的差异表达基因,并揭示其在生物学功能、分子过程和细胞组分方面的富集情况,为后续的生物学实验和机制研究提供理论依据。
实验步骤:1.基因芯片数据的预处理:包括数据导入、数据清洗、标准化和基因注释等。
首先,将基因芯片数据导入到数据分析软件中,然后针对数据质量进行清洗,剔除异常值和低质量的基因。
接下来,对基因表达谱数据进行归一化处理,保证不同芯片之间的数据可比性。
最后,对基因进行注释,将基因名与其对应的功能注释进行关联。
2.差异分析:通过比较不同组别之间的基因表达差异,筛选出差异表达基因。
差异分析方法包括t检验、方差分析等。
根据统计学中的显著性水平,设定p值的阈值,将差异表达基因筛选出来。
3.聚类分析:将差异表达基因按照其表达谱进行聚类分析,可将具有相似表达模式的基因聚集在一起。
常用的聚类方法包括层次聚类和K均值聚类等。
实验结果与分析:通过基因芯片数据处理和分析,我们得到了与研究目标相关的差异表达基因。
结合GO分析的结果,我们可以进一步了解这些差异表达基因在生物学功能、分子过程和细胞组分方面的富集情况。
例如,在生物学过程方面,我们可以得知这些基因是否与细胞增殖、凋亡、信号传导等生物学过程相关;在分子功能方面,我们可以了解这些基因是否具有催化活性、结合能力等分子功能特性;在细胞组分方面,我们可以了解这些基因在细胞核、细胞质、细胞膜等细胞组分的分布情况。

基因组学研究中的表达谱数据分析实验报告1. 研究背景和目的基因组学研究是一门研究基因组结构、功能和表达等方面的学科。
其中,表达谱数据分析是基因组学研究中重要的一环,它可以帮助我们了解基因在不同条件下的表达情况,从而揭示基因调控和生物过程等方面的机制。
本实验的目的是利用表达谱数据分析方法,解析某组织或细胞在不同条件下的基因表达谱谱,以及评估基因的差异表达情况和通路富集分析。
2. 数据获取和预处理我们首先获得了一组某种生物体在不同处理条件下的表达谱数据。
这些数据可以通过RNA测序等技术获得,其中包含了上千个基因的表达水平信息。
在进行数据分析之前,我们需要对原始数据进行预处理。
首先,我们对原始测序数据进行质控,剔除低质量的碱基和低测序深度的样本。
随后,我们利用比对算法将测序reads与参考基因组序列进行比对,得到每个基因的计数信息。
最后,我们对每个基因的计数进行归一化处理,通过计算fpkm或tpm等指标,将其转化为相对表达水平。
3. 差异表达基因分析在基因表达谱的分析中,我们通常关注基因在不同样本中的表达水平差异。
为了找出差异表达基因,我们采用了差异表达分析方法,如DESeq2、edgeR等。
在差异表达分析中,我们根据各个基因的表达水平,采用统计模型来计算差异表达的显著性。
通过比较不同处理条件下的样本,我们可以得到差异表达基因的列表。
通过差异表达基因分析,我们可以发现与不同处理条件相关的基因,并进一步探究其与生物学过程、通路的关联。
4. 通路富集分析为了更好地理解差异表达基因的生物学功能和调控机制,我们进行了通路富集分析。
通路富集分析可以帮助我们找到与差异表达基因相关的生物通路和功能模块。
在通路富集分析中,我们将差异表达基因与已知的生物通路数据库进行比对,利用统计方法来判断差异表达基因在某个通路中的富集程度的显著性。
通过这种方法,我们可以识别出与特定处理条件相关联的通路和功能模块,为进一步的研究提供线索。

基因表达谱分析方法的使用教程基因表达谱分析是研究基因在细胞或组织中的表达水平及其变化的重要手段。
通过分析基因表达谱,可以揭示基因在不同生理状态和疾病发生发展过程中的作用机制,为进一步的研究提供重要依据。
本文将介绍几种常用的基因表达谱分析方法及其使用教程。
1. microarray芯片技术Microarray技术是目前最常用的基因表达谱分析技术之一。
它通过在玻璃片或硅片上固定大量的探针序列,然后将待测样品中的RNA反转录成cDNA标记,与芯片上的探针序列杂交,利用荧光探针检测标记的cDNA的信号强度来反映基因的表达水平。
使用microarray技术进行基因表达谱分析的步骤如下:1) 设计实验:确定实验的目的、样品来源、实验方案和所需探针等。
2) 样品处理:提取RNA并进行反转录,将RNA转化为cDNA,并标记荧光。
3) 芯片处理:将标记的cDNA杂交于芯片上的探针序列,完成杂交后进行芯片洗涤。
4) 扫描芯片:使用芯片扫描仪检测芯片上标记的cDNA的荧光信号。
5) 数据分析:对扫描得到的图像进行图像分析、标准化、差异基因筛选和功能富集分析等。
2. RNA测序技术RNA测序技术(RNA-Seq)是一种基于高通量测序平台的基因表达谱分析技术,它通过直接测序RNA分子,可以定量测量每个基因的表达水平,并发现新基因和变异。
使用RNA测序技术进行基因表达谱分析的步骤如下:1) RNA提取:从细胞或组织中提取RNA,并进行质量检测和纯化。
2) RNA片段化和逆转录:将RNA经过碱水解或酶处理,得到短的RNA 片段,然后进行逆转录合成cDNA。
3) 文库构建:对cDNA进行末端修复、A尾化、连接测序接头和PCR扩增等处理,构建测序文库。
4) 文库测序:将测序文库装载到测序仪上,进行高通量测序,得到原始测序数据。
5) 数据分析:对原始测序数据进行质控、对齐、定量、差异基因表达分析以及功能注释等。
3. qRT-PCR技术量子链反应实时荧光定量聚合酶链式反应(qRT-PCR)是一种常用的基因表达谱分析方法,其优点在于准确、灵敏和高通量。
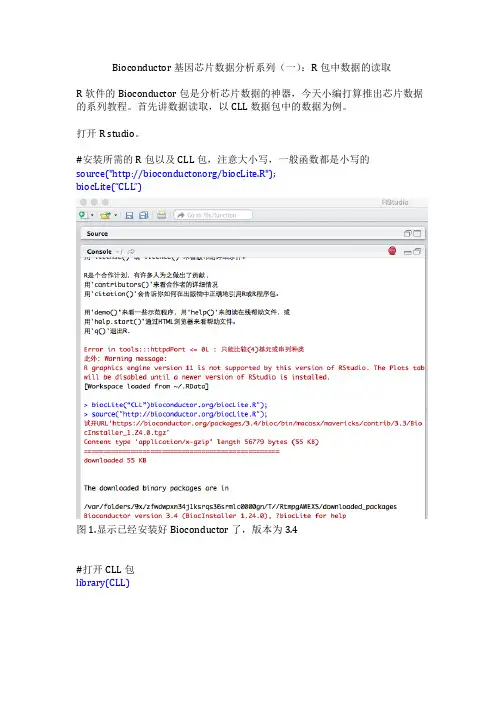
Bioconductor基因芯片数据分析系列(一):R包中数据的读取R软件的Bioconductor包是分析芯片数据的神器,今天小编打算推出芯片数据的系列教程。
首先讲数据读取,以CLL数据包中的数据为例。
打开R studio。
#安装所需的R包以及CLL包,注意大小写,一般函数都是小写的source("/biocLite.R");biocLite(“CLL”)图1.显示已经安装好Bioconductor了,版本为3.4#打开CLL包library(CLL)图2.显示打开CLL成功图3.右侧栏内可见看到目前载入的程序包data(CLLbatch)#调用RMA算法对数据预处理CLLrma<-rma(CLLbatch)#读取处理后所有样品的基因表达值e<- exprs(CLLrma)#查看数据e我们可以看到,CLL数据集中共有24个样品(CLL10.CEL, CLL11.CEL, CLL12.CEL, 等),此数据集的病人分为两组:稳定组和进展组,采用的设计为两组之间的对照试验(Control Test)。
从上面的结果可知,Bioconductor具有强大的数据预处理能力和调用能力,仅仅用了6行代码就完成了数据的读取及预处理。
Bioconductor基因芯片数据分析系列(二):GEO下载数据CEL的读取首先得下载一个数据,读取GEO的CEL文件采用如下命令:登陆pubmed,找到一个你感兴趣的数据库在底下栏目下载CEL文件打开R软件#安装所需的R包以及CLL包,注意大小写,一般函数都是小写的source("/biocLite.R");biocLite(“CLL”)>library(affy)>affybatch<- ReadAffy(celfile.path = "GSE36376_RAW")请注意目录的路径,在window下,反斜杠‘\’要用转义字符“\\”表示。
⼀款表达谱数据分析的神器--CCLE--转载现在做⽣物和医学的,很多都可能会和各种组学数据打交道。
其中表达谱数据总是最常⽤的,也是⽐较好测的。
即使在⼯作中不去测序,也可以利⽤已有的数据库去做⼀些数据挖掘,找⼀找不同表型(⽐如癌症)对应的marker或者调控的通路。
这⾥跟⼤家推荐⼀款分析表达谱数据的神器GENE-E,亲测⽐较好⽤,满⾜常⽤需求,可以做各种热图,聚类,箱图,相似分析和标记筛选等等,⽽且⾮常⼩巧。
进⼊软件之后,界⾯如下,这⾥显⽰的就是基因表达的热图(heatmap),其中蓝⾊表⽰被低调控,红⾊表⽰被⾼调。
横向是不同的细胞系,纵向是不同的基因和探针名。
数据操作第⼀次进⼊程序可以利⽤它⾃带的测试数据来上⼿,如下图,打开File->OpenExample Data-> CCLE,然后需要⼏分钟来下载数据,别忘记连⽹。
CCLE是Board Institute维护的⼀个癌症细胞系的数据库,⽬前有近千个cell line,有兴趣的也可以去官⽹看看。
等⼏分钟load完数据之后了就可以挑选感兴趣的数据进⾏分析了。
⽐如可以在圆圈处敲⼊skin ovary,即挑选和⽪肤癌以及卵巢癌有关的细胞系(此时带有关键词的列名已经被橙⾊⾼亮)。
然后点击⿊⾊箭头处的图标,既可以选中这些列。
然后点击图中图标,建⽴新的热图(heatmap)。
相似分析对两种癌症的数据就可以进⾏进⼀步分析。
⽐如我们可以分析不同细胞系间的相似性,如下图Tools->Similarity Matrix,点击ok即可。
得到相似性如下图(这⾥为了计算更快,只取了其中部分数据),其中矩阵中的(i,j)位置对应细胞系i和细胞系j之间的相似性,红⾊表⽰相似度⾼。
矩阵上⽅和右⽅的⾊条表⽰的是卵巢癌(深蓝)或⽪肤癌(浅蓝)的细胞系。
我们可以发现⼀个位置来源的癌细胞都相互之间很像。
聚类分析另外我们也可以做聚类分析,如下图,点击图标后输⼊参数。
这⾥需要选择对列(Column)还是⾏(Row)聚类。
基因表达芯片数据的预处理和分析基因表达芯片是一种目前广泛应用于生物医学研究中的技术,它可以帮助研究人员在分子水平上对细胞、组织、器官及其疾病发生机制进行深入研究,从而为疾病的诊断、治疗和药物研发等领域提供有力的支持。
基因表达芯片所涉及的数据处理步骤较多,其中预处理和分析是其中最为基础和关键的两个环节。
本文将从这两个方面详细阐述基因表达芯片数据的预处理和分析。
一、基因表达芯片数据的预处理预处理部分主要包括质量控制、数据归一化和拼接等步骤。
具体介绍如下:1、质量控制质量控制是基因表达芯片数据预处理中非常重要的一步,它的目的是检查芯片实验结果的质量。
通过质量控制可以发现数据中的异常现象,包括低质量的样品、芯片实验中的坏控制等。
一旦发现问题,需要对其进行相应的策略处理,以确保测量结果的正确性和准确性。
2、数据归一化数据归一化是指将不同富集度的探测物本底进行标准化处理,以能够在同一芯片上比较不同样品的水平。
目前普遍使用的归一化方法有MAS5、RMA、GCRMA 和Ebtiseh等。
其中MAS5方法独立于信号内容以及噪声分布,不需要对数据做任何假设。
RMA方法适用于多共同贡献的基因表达的依赖性模型。
GCRMA方法基于模型的切断比值方法,可以有效消除芯片噪声的影响。
Ebtiseh方法可以充分利用芯片的信息,并通过最佳阈值确定最佳归一化方案。
3、拼接拼接是指将一组芯片测量数据进行合并,形成一个较大的数据矩阵。
拼接的目的是将不同个体、不同时间点的基因表达芯片测量结果进行统一处理,为后续的差异分析和数据挖掘提供支持。
二、基因表达芯片数据的分析基因表达芯片数据分析主要包括差异分析、功能分析和网络分析等步骤。
具体介绍如下:1、差异分析差异分析是指比较两组或多组样品之间的基因表达水平差异。
差异分析的主要方法有t检验、方差分析、多重比较法、基因表达芯片的类别分析以及机器学习算法。
通过差异分析可以找到与疾病有关的不同表达基因。
芯片项目分析内容说明(示例):
1)原始芯片数据处理:
我们重老师提供的数据列表中下载了408张非重复的芯片,这些芯片来自13批不同的数据,
首先,我们使用RMA算法对每一个批次的芯片,分批进行了信号值处理,经过PCA分析后
发现,不同的芯片按照不同的批次被分开,说明来自不同实验的数据之间存在非常强的批次
效应。
对应文件为: 01原始芯片数据处理\RMA_in_Batches\all_exp.xls.PCA3D.pdf
截止,我们将所有芯片的放在一起,一起使用RMA算法进行信号值处理,经过PCA分析后
发现,纵使是将所有芯片一起进行RMA处理,这些来自不同实验的数据还是存在批次效应。
对应文件为:01原始芯片数据处理\RMA_in_Batches\ all_exp.xls.PCA3D.pdf
2)批次效应矫正:
因为,来自不同实验的数据之间存在批次效应,所以我们使用基于经验贝叶斯方法的ComBat
算法对不同批次的数据进行批次效应矫正。
我们使用将全部芯片放在一起进行RMA处理后的数据作为输入文件,进行批次效应表达量
矫正,然后使用PCA分析发现,批次效应基本被消除掉了。
对应文件为:02批次效应矫正\校正前\all_exp.xls.PCA3D.pdf
02批次效应矫正\校正后\Adjusted.all_exp.xls.out.xls.out.PCA3D.pdf
3)差异分析:
我们使用t_test和方差分析对批次效应矫正前后的数据都进行差异检测,我们使用t_test的
pvalue<0.01和方差分析的pvalue<0.01为标准选取差异基因,对于校正后我们共得到26760
个差异探针,对于校正前我们23349个差异探针,我们对得到的差异探针都经行了PCA分
析和cluster分析
对应文件:03差异表达\
4)特征选择:
我们使用SVMRFE算法,一种基于支持向量机的特征选取算法,对差异探针,进行了特诊选
择。我们对校正前和校正后的差异探针都进行了特征选择
SVMRFE会将特征基因按照从高到低的顺序进行排序,我们选择排名前10,前20,前30,前
40的探针,进行PCA分析,观测使用特征基因进行分类是否准确。
对应文件:04SVMRFE\
其中nohup.out文件为进行SVM分类时,选取不同数量特征基因,使用留一法交叉验证所
得到的准确率。
下图为特征选择前后样本的热图分析结果。
SVM分析前(2562 genes) SVM分析后(19 genes)
5)异常样本去除:
我们使用PCA分析得到样本在PC1,PC2,PC3上的位置,然后计算每个样本在3个PC上与
其均值的距离的平方,然后分别乘以其PC权重后求和,然后求z score,对求得的zscore值
进行高斯累积分布,然后将值大于0.8的样本作为离群样本,去除掉。对于肿瘤样本,和正
常样本我们分别计算各自的离群样本。
对于批次校正前的数据,我们得到癌症的离群样本为41,正常的离群样本为7
对于其次矫正后的数据,我们得到癌症的离群样本为49,正常的离群样本为7
对应文件夹:05离群样本检测_PCA_Gaussian_cumulative_distribution/
下图为3D PCA结果(例图)
6)去除异常样本后重新进行SVMRFE:
我们将异常样本去除后,又重新进行了SVMRFE特征选择。
结果目录:06去掉离群样本/
7)批次校正对结果影响的评估
从结果来看,不使用批次矫正得到结果更优,其原因是因为,本次芯片数据时正常和肿瘤样
本的数量极度不对称,且有些批次的数据全部为肿瘤样本,在此种情况下进行批次矫正会产
生过矫正。即将肿瘤样本的特征,当做批次特征,进行矫正,这种情况下即产生了过矫正效
应,不能反应样本真实情况。建议使用批次校正前的数据进行后面的参数优化
8)异常样本去除参数优化:
之前我们使用PCA+高斯累计分布,使用参数0.8的时候,
对于批次校正前的数据,我们得到癌症的离群样本为41,正常的离群样本为7。
后面我们调整了这个参数的设置,使用参数0.95的时候,
对于批次校正前的数据,我们得到癌症的离群样本为16,正常的离群样本为7。
后面我们在此调整了这个参数的设置,使用参数0.99的时候,
对于批次校正前的数据,我们得到癌症的离群样本为12,正常的离群样本为3。
从结果看使用0.99为参数去掉异常样本后分析的结果会出现错分的情况,而使用0.95为参
数则没有出现错分的情况。