砷和铅的测定方法
- 格式:docx
- 大小:17.17 KB
- 文档页数:3
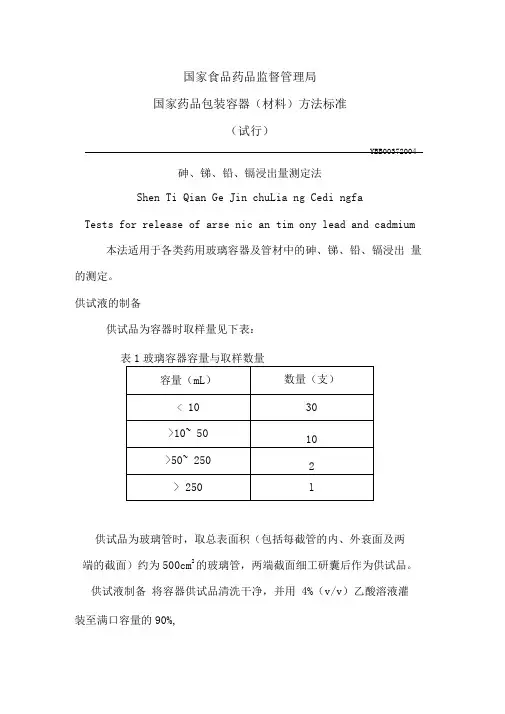
国家食品药品监督管理局国家药品包装容器(材料)方法标准(试行)YBB00372004砷、锑、铅、镉浸出量测定法Shen Ti Qian Ge Jin chuLia ng Cedi ngfaTests for release of arse nic an tim ony lead and cadmium 本法适用于各类药用玻璃容器及管材中的砷、锑、铅、镉浸出量的测定。
供试液的制备供试品为容器时取样量见下表:表1玻璃容器容量与取样数量供试品为玻璃管时,取总表面积(包括每截管的内、外衰面及两端的截面)约为500cm2的玻璃管,两端截面细工研囊后作为供试品。
供试液制备将容器供试品清洗干净,并用4%(v/v)乙酸溶液灌装至满口容量的90%,对于安瓶瓿等容量较小的容器,则灌装乙酸溶液至瓶身缩肩部.用倒置烧杯(需用平均线热膨胀系数a (20C〜300C)约为3.3X 10-6K^1硼硅玻璃制成,新的烧杯须经过老化处理)或惰性材料铝箔盖住口部。
98C蒸煮2小时。
冷却后取出供试品,溶液即为供试液。
将玻管供试品清洗干净,置入装有4%(v/v)乙酸溶液1000mL的玻璃容器(玻璃容器不应含有砷、锑、铅、镉元素)中,98C蒸煮2 小时.冷却后取出供试品,溶液即为供试液。
1砷浸出量测定法试验原理供试液中含有的高价砷被碘化钾、氯化亚锡还原为三价砷.然后与锌粒和酸反应产生的新生态氢,生成砷化氢,经银盐溶液吸收后,形成红色胶态物,与标准曲线比较,测定其含量。
测定法精密量取供试液10mL、空白液10mL、标准砷溶液(每1 mL 相当于I 卩g 的As) 1 mL、2mL、3 mL、4 mL、5 mL (必要时可根据样品实际情况调整线性范围),分别置测砷瓶中,按中华人民共和国药典2000年版二部附录忸J砷盐检查法第二法操作,用分光光度法,在510nm的波长处测定吸收度。
以浓度为X轴,以吸收度为Y 轴,绘制标准曲线.与标准曲线比较确定供试品的浓度。

土壤质量总汞总砷总铅的测定原子荧光法土壤质量是影响农作物生长和环境保护的重要指标之一。
土壤中重金属元素的含量是评价土壤质量的关键因素之一。
其中,总汞(Total mercury, THg)、总砷(Total arsenic, TAs)和总铅(Total lead, TPb)是对土壤环境质量进行评估的重要指标。
为了测定土壤中这些重金属元素的含量,常采用原子荧光法进行分析。
原子荧光法是一种基于原子吸收、发射或荧光原理的分析方法,适用于各种样品中重金属元素的测定。
这种方法具有灵敏度高、选择性强、操作简便和多元素同时分析的优点,因此广泛应用于土壤、水体、植物等环境样品的分析。
在土壤中,总汞、总砷和总铅的测定需要经过样品的前处理、原子化和检测等步骤。
首先,样品的前处理对土壤样品进行干燥、研磨、筛选等处理,以去除杂质,提高分析的准确性和灵敏度。
土壤样品通常通过干燥箱或真空烘箱进行干燥,然后使用球磨机等设备对土壤进行研磨,最后通过不同孔径的筛网进行筛选,得到符合要求的土壤粉末样品。
接下来,将土壤样品中的重金属元素原子化。
常用的原子化方法有火焰原子吸收法(Flame Atomic Absorption Spectrophotometry, FAAS)、电感耦合等离子体质谱法(Inductively Coupled Plasma Mass Spectrometry, ICP-MS)等。
其中,ICP-MS方法具有高灵敏度、高选择性和多元素同时测定的优点,被广泛应用于土壤重金属元素的分析。
最后,通过原子荧光光谱仪对土壤样品中的重金属元素进行检测。
原子荧光光谱仪是一种专用仪器,通过激发样品中的重金属元素原子,使其发射荧光信号,然后通过对荧光信号的测量和分析,确定重金属元素的含量。
原子荧光光谱仪具有高分辨率、高稳定性和高精确度的特点,能够准确测定样品中微量重金属元素的含量。
总的来说,土壤质量中总汞、总砷和总铅的测定主要采用原子荧光法进行分析。

ICP-OES法快速测定化妆品中铅、砷、汞摘要:化妆品试样经硝酸消解,将各种元素溶出到酸溶液中,使用电感耦合等离子体发射光谱仪测定样品中的铅,砷,汞。
该方法定量检出限为铅:2 mg/L、砷 1 mg/L、汞0.2 mg/L。
线性相关系数≥0.995。
在精华水、精华露和粉饼中的元素添加回收率在85%~115%之间、精密度<15%。
符合日常快速检测的要求。
关键词:等离子发射光谱化妆品铅砷汞化妆品中铅,砷,汞等元素是主要污染元素,是进出口化妆品的主要检测项目。
根据《化妆品卫生规范》 2007年版的规定,化妆品中铅不得超过40 mg/kg;砷不得超过10 mg/kg;汞不得超过1 mg/kg。
目前,测量化妆品中元素的方法主要包括有《化妆品卫生规范》 2007年版第三部分规定的火焰原子吸收法、氢化物原子荧光法等方法。
这些方法都是比较常用的方法,虽能满足检测要求,但也有一定的局限性,主要是高通量能力不足,不能满足实验室大量化妆品样品检测的要求。
所以,根据上述限量要求和本实验室的检测经验,实验室制定了一次消解,采用等离子发射光谱法同时测量铅、砷、汞的检测方法,大大提高了检测效率。
1 实验部分1.1 仪器和试剂电感耦合等离子体发射光谱仪(PE OPTIMA8000DV,美国PE公司);消解仪、水浴锅或类似加热装置,加热的最小温度为100℃。
超纯水:18MΩcm;硝酸,优级纯或经过提纯;1.2 标准溶液铅、砷标准溶液:有证标准溶液。
临用时逐级稀释,先配成铅20mg/L,砷10mg/L 的混合储备溶液,配置在5%硝酸中,使用期为三个月。
临用时逐级稀释,最终配成铅浓度为40 μg/L,80 μg/L,120 μg/L,160 μg/L,200 μg/L,砷浓度为20 μg/L,40 μg/L,60 μg/L,80 μg/L,100 μg/L的混合标准工作液,配置在2%硝酸中,使用期为一个月。
汞标准溶液:有证标准溶液。
1.目的:建立铅、镉、砷、汞、铜测定法操作规程,规范铅、镉、砷、汞、铜测定法的操作。
2.范围:本公司产品铅、镉、砷、汞、铜的检验。
3.责任:QC检验员。
4.内容:4.1 原子吸收分光光度法:本法系采用原子吸收分光光度法(附录Ⅴ D)测定中药材中的铅、镉、砷、汞、铜,除另有规定外,按下列方法测定。
4.1.1铅的测定(石墨炉法)4.1.1.1测定条件4.1.1.1.1参考条件:波长 283.3nm,干燥温度 100~120℃,持续 20 秒;灰化温度 400~750℃,持续 20~25 秒;原子化温度 1700~2100℃,持续 4~5秒;背景校正为氘灯或塞曼效应。
4.1.1.2铅标准储备液的制备精密量取铅单元素标准溶液适量,用 2%硝酸溶液稀释,制成每 1ml 含铅(Pb)1µg 的溶液,即得(0~5℃贮存)。
4.1.1.3标准曲线的制备分别精密量取铅标准储备液适量,用 2%硝酸溶液制成每1ml 分别含铅 0ng、5ng、20ng、40ng、60ng、80ng 的溶液。
分别精密量取 1ml,精密加含 1%磷酸二氢铵和 0.2%硝酸镁的溶液 1ml,混匀,精密吸取20µl 注入石墨炉原子化器,测定吸光度,以吸光度为纵坐标,浓度为横坐标,绘制标准曲线。
4.1.1.4供试品溶液的制备4.1.1.4.1 A 法取供试品粗粉 0.5g,精密称定,置聚四氟乙烯消解罐内,加硝酸3~5ml,混匀,浸泡过夜,盖好内盖,旋紧外套,置适宜的微波消解炉内,进行消解(按仪器规定的消解程序操作)。
消解完全后,取消解内罐置电热板上缓缓加热至红棕色蒸气挥尽,并继续缓缓浓缩至2~3ml,放冷,用水转入 25ml 量瓶中,并稀释至刻度,摇匀,即得。
同法同时制备试剂空白溶液。
4.1.1.4.2 B 法取供试品粗粉 1g,精密称定,置凯氏烧瓶中,加硝酸高氯酸(4∶1)混合溶液 5~10ml,混匀,瓶口加一小漏斗,浸泡过夜。

GB/T22105.1-2008土壤质量总汞、总砷、总铅的测定原子荧光法1范围GB/T22105的本部分规定了土壤中总汞的原子花光光谱测定方法。
本部分适用于土壤中总汞的测户定。
本部分方法检出限0.002mg/kg。
2原理采用硝酸-盐酸混合试剂在沸水浴中加热消解土壤试样,再用硼氢化钾(KBH4)或硼氢化钠(NaBH4)将样品中所含汞还原成原子态汞,由载气(氩气)导入原子化器中在特制汞空心阴极灯照射下,基态汞原子被激发至高能态,在去活化回到基态时,发射出特征波长的荧光,其荧光强度与汞的含量成正比。
与标准系列比较,求得样品中汞的含量。
3试剂本部分所使用的试剂除另有说明期外,均为分析纯试剂,试验用水为去离子水。
3.1盐酸(HCL),p=1.19g/ml,优级纯。
3.2硝酸(HNO3)p=1.42g/ml,优级纯。
3.3硫酸(H2SO4),p=1.84g/ml,优级纯。
3.4氢氧化钾(KOH),优级纯。
3.5硼氢化钾(KBH4),优级纯。
3.6重铬酸钾(K2Cr5O7),优级纯。
3.7氯化汞(HgCl2),优级纯。
3.8硝酸盐酸混合试剂[(1+1)王水]:取1份硝酸(3.2)与3份盐酸(3,1)混合,然后用去离子水稀释一倍。
3.9还原剂[0.01%硼氢化钾(KBH4)+0.2%氢氧化钾(KOH溶液]称取0.2g氢氧化钾(3.4)放入烧杯中,用少量水溶解,称取0.01g硼氢化钾(3.5)放入氢氧化钾溶液中,用水稀释至100mL,此溶液现用现配。
3.10载液[(1+19)硝酸溶液]:量取25mL硝酸(3.2),缓缓倒入放有少量去离子水的500mL容量瓶中,用去离子水定容至刻度,摇匀。
3.11保存液:称取0.5g重铬酸钾(3.6),用少量水溶解,加入50mL硝酸(3.2),用水稀释至1000mL,摇匀。
3.12稀释液:称取0.2g重铬酸钾(3.6),用少量水溶解,加入28mL硫酸(3.3),用水稀释至1000mL,摇匀。

碘量法硫化物监测方法采用碘量法监测1.方法原理硫化物在酸性条件下,与过量的碘作用,剩余的碘用硫代硫酸钠溶液滴定。
由硫代硫酸钠溶液所消耗的量,间接求出硫化物的含量。
2.干扰及消除还原性或氧化性物质干扰测定。
水中悬浮物或浑浊度高时,对测定可溶态硫化物有干扰。
遇此情况应进行适当处理。
3.方法的适用范围本方法适用于含硫化物在1mg/L以上的水和废水的测定。
4.仪 器4.1 250ml碘量瓶。
4.2 中速定量滤纸或玻璃纤维滤膜。
4.3 25ml或50ml滴定管(棕色)。
5.试 剂5.1 1mol/L乙酸锌溶液:溶解220g乙酸锌于水中,用水稀释到1000ml。
5.2 1%淀粉指示液。
5.3 1+5mol/L硫代硫酸钠标准溶液:称取12.4g硫代硫酸钠溶于水中,稀释至1000ml,加入0.2g无水硫酸钠,保存于棕色瓶中。
标定:向250ml碘量瓶内,加入1g碘化钾及50ml水,加入的重铬酸钾标准溶液15.00ml,加入5+1硫酸5ml,密塞混匀。
置暗处静置5min,用待标定的硫代硫酸钠标准溶液滴定至溶液淡黄色时,加入1ml淀粉指示液,继续滴定至蓝色刚好消失,记录标准液用量(同时作空白滴定)。
硫代硫酸钠标准溶液的浓度按下式计算:c=15.00/(V1-V2)×0.05式中,V1――滴定重铬酸钾标准溶液消耗硫代硫酸钠标准溶液体积(ml);V2――滴定空白溶液消耗硫代硫酸钠标准溶液体积(ml);0.05――重铬酸钾标准溶液的浓度(mol/L)。
6.步 骤将硫化锌沉淀连同滤纸转入250ml碘量瓶中,用玻璃棒搅碎,加50ml水及10.00ml碘标准溶液,5ml1+5硫酸溶液,密塞混匀。
暗处放置5min,用硫代硫酸钠标准溶液滴定至溶液呈淡黄色时,加入1ml淀粉指示液,继续滴定至蓝色刚好消失,记录用量。
同时作空白试验。
水样若经酸吹气预处理,则可在盛有吸收液的原磺量瓶中,同上加入试剂进行测定。
7.计 算硫化物=(V0-V1)c×16.03×1000/V式中:V0――空白试验中,硫代硫酸钠标准溶液用量(ml);V1――水样滴定时,硫代硫酸钠标准溶液用量(ml);V――水样体积(ml);16.03――硫离子摩尔质量(g/mol);c――硫代硫酸钠标准溶液浓度(mol/L)。

的测定土壤质量 总砷测定 原子荧光法(GB/T 22105.2-2008)方法确认报告1. 目的通过原子荧光法测定土壤中砷的检出限、精密度、准确度,加标回收率,来判断本实验室此方法是否合格。
2. 适用范围及方法标准依据方法依据:GB/T 22105.2-2008本标准适用于土壤和沉积物中总砷的测定。
本方法检出限为 0.01mg/kg。
3.方法原理样品中的砷经加热消解后,加入硫脲使五价砷还原为三价砷,再加入硼氢化钾将其还原为砷化氢,由载气(氩气)导入原子化器进行原子化分解为原子态砷,在特制砷空心阴极灯的发射光激发下产生原子荧光,其荧光强度与试样中砷的含量成正比。
与标准系列比较,求得样品中汞的含量。
4 试剂和材料除非另有说明,分析时均使用符合国家标准的分析纯试剂,实验用水为去离子水。
4.1 盐酸(HCl):ρ=1.19 g/ml。
4.2 硝酸(HNO3):ρ=1.42 g/ml。
4.3 氢氧化钾(KOH):优级纯。
4.4 硼氢化钾(KBH4):优级纯。
4.5 硫脲(CH4N2S):分析纯。
4.6 抗坏血酸(C6H8O6):分析纯。
4.7 三氧化二砷(As2O3):优级纯。
4.8 王水(1+1):取1份硝酸(4.2)与3份盐酸(4.1)混合,然后用去离子水稀释一倍。
4.9 还原剂:称取 0.2g 氢氧化钾(4.3)放入盛有 100 ml 蒸馏水的烧杯中,玻璃棒搅拌待完全溶解后再加入称好的 1.0g 硼氢化钾(4.4),搅拌溶解。
此溶液当日配制,用的测定于测定砷。
4.10 载液 1+9盐酸溶液:量取50ml盐酸(4.1),缓缓倒入放有少量去离子水的500ml 容量瓶中,用去离子水定容至刻度,摇匀。
4.11 硫脲溶液(5%):称取10g硫脲(4.5)溶解于200ml水中,摇匀。
用时现配。
4.12 抗坏血酸(5%):称取10g抗坏血酸(4.6)溶解于200ml水中,摇匀。
用时现配。
4.12 砷标准储备液:ρ=1.00mg/mL采用从环境保护部标准样品研究所购买的砷标准储备液4.13 金属标准使用溶液4.13.1 砷标准使用液:ρ= 1.00μg/mL用(1+9)盐酸溶液(4.10)稀释砷标准贮备液10倍,然后再稀释100倍得;5 仪器和设备5.1 氢化物发生原子荧光光度计。

土壤中重金属测定国标一、对于土壤中重金属的测定,应按照GB 15618-1995《土壤环境中重金属污染物危害防治标准》(以下简称“标准”)中所规定的方法进行测定。
二、土壤中重金属元素的测定,包括有铅、汞、镉、砷、铬、铜、锌、镍、铁元素,应按照标准中所规定的方法确定,其中铅、汞、镉、砷测定方法如下:1、铅、汞、镉、砷测定:(1)样品制备:土壤样品要求按照GB 4789.1-1997《食品安全微生物学检验密闭械法检验程序》中第7.2.2节规定的方法消毒制备,采用活性炭净化法提取土壤中砷、镉、铅和汞,提取条件和提取物稀释方法按照标准中的要求,提取物接近浓缩。
(2)重金属元素测定:采用气相色谱质谱联用(GC/MS)的方法,确定砷、镉、铅和汞的浓度,具体的操作方法和水平如下:(a)石英柱温度要求:程序从60℃->70℃->80℃,步长为10℃,时间为3min;(b)检测气相吸收剂:以苯、苯乙烯作为检测气体;(c)光机:采用铱钌灯,电压32V,电流200mA;(d)重金属元素测定水平:铅(Pb)20-400 mg/kg,汞(Hg)2-50 mg/kg,镉(Cd)2-50mg/kg,砷(As)0.5-50mg/kg。
三、根据标准规定,《土壤环境中重金属污染物危害防治标准》对土壤中重金属元素的各项指标进行了规定。
重金属元素含量按GB15619-1995标准中允许的土壤环境限量值来衡量,铅(Pb)400 mg/kg、汞(Hg)50 mg/kg、镉(Cd)50 mg/kg、砷(As)50mg/kg,超出该规定则视为重金属元素污染。
四、在测定土壤中重金属元素时,应严格按照标准的规定进行测定。
操作中一定要学习正确的技术,并严格遵守操作要求;样品的采集、制备以及污染物的提取都很重要,尤其是土壤的消毒;气相色谱质谱联用仪器的使用和调试也很重要,要掌握其使用技术;最后,根据标准的要求来准确测定和判定,严格控制其质量,以确保土壤环境的安全和健康。

方法验证报告土壤砷铬镉铅和镍的测定土壤中的砷(As)、铬(Cr)、镉(Cd)、铅(Pb)和镍(Ni)等重金属元素,是由于人类活动和自然灾害造成的一种环境污染。
这些重金属元素在土壤中的积累会对生态系统和人体健康产生潜在的风险,因此对土壤中这些重金属元素的测定尤为重要。
本文将介绍一种常用的方法验证报告,用于测定土壤中砷、铬、镉、铅和镍的含量。
1.实验目的本次实验的目的是验证一种方法用于测定土壤中砷、铬、镉、铅和镍元素的含量。
2.实验原理本实验使用的方法是原子吸收光谱法(AAS)。
原子吸收光谱法是一种常用的重金属元素的分析方法,基于原子的吸收光谱特性。
在实验中,土壤样品首先经过适当的前处理步骤,如提取和预处理等,然后用AAS仪器进行测定。
在AAS仪器中,样品中的重金属元素被蒸发和原子化,然后通过原子吸收光谱分析。
3.实验步骤a.样品的前处理:取适量土壤样品(约10g),加入足量盐酸(HCl),进行酸溶解。
然后,对溶解液进行过滤,获得清澈的溶液。
b.原子吸收光谱测定:将溶液转移到AAS仪器中,根据仪器的操作说明进行测定。
根据实验需要,可以选择不同的光谱线进行测定。
c.标准曲线的绘制:准备一系列浓度已知的标准溶液,分别进行AAS测定。
然后,根据测定结果绘制标准曲线,以便后续计算目标元素的含量。
4.数据处理a.计算目标元素的含量:根据实验测定结果和标准曲线,可以计算出样品中目标元素的含量。
根据实验需要,可以选择不同的计算公式进行计算。
b.数据统计和分析:对实验测定结果进行统计和分析,包括计算平均值、标准差等,以评估实验结果的准确性和可靠性。
5.结果和讨论在实验中得到了土壤样品中砷、铬、镉、铅和镍元素的测定结果。
根据实验的目的和要求,可以对结果进行分析和讨论,如比较不同样品的含量差异、评估土壤中重金属元素的污染程度等。
6.结论根据实验结果和讨论,可以得出关于样品中砷、铬、镉、铅和镍元素含量的结论。
根据需要,可以进一步提出改进方法的建议,以提高测定的准确性和可靠性。

食品中铅的测定:第一法石墨炉原子吸收光谱法3原理试样经灰化或酸消解后,注入原子吸收分光光度计石墨炉中,电热原子化后吸收283.3 nm共振线,在一定浓度范围,其吸收值与铅含量成正比,与标准系列比较定量。
4试剂和材料硝酸:优级纯。
4.2过硫酸铵。
4.3过氧化氢(30% )。
4.4高氯酸:优级纯。
4.5硝酸(1 + 1):取50 mL硝酸慢慢加入50 mL水中。
4.6硝酸(0.5 mol/L ):取3.2 mL硝酸加入50 mL水中,稀释至100 mL。
4.7硝酸(I mo1/L):取6.4 mL硝酸加入50 mL水中,稀释至100 mL。
4.8磷酸二氢铵溶液(20 g/L ):称取2.0 g磷酸二氢铵,以水溶解稀释至100 mL。
4.9混合酸:硝酸十高氯酸(9+ 1)。
取9份硝酸与1份高氯酸混合。
4.10铅标准储备液:准确称取1.000 g金属铅(99.99%),分次加少量硝酸(4.5),加热溶解,总量不超过37 mL ,移入1000 mL容量瓶,加水至刻度。
混匀。
此溶液每毫升含1.0 mg 铅。
4.11铅标准使用液:每次吸取铅标准储备液 1.0 mL于100 mL容量瓶中,加硝酸(4.6)至刻度。
如此经多次稀释成每毫升含10.0 ng, 20.0 ng, 40.0 ng, 60.0 ng, 80.0 ng铅的标准使用液。
5仪器和设备5.1原子吸收光谱仪,附石墨炉及铅空心阴极灯。
5.2马弗炉。
5.3天平:感量为1 mg。
5.4干燥恒温箱。
5.5瓷坩埚。
5.6压力消解器、压力消解罐或压力溶弹。
5.7可调式电热板、可调式电炉。
6分析步骤6.2试样消解(可根据实验室条件选用以下任何一种方法消解)6.2.1湿式消解法:称取试样 1 g〜5 g (精确到0.001 g )于锥形瓶或高脚烧杯中,放数粒玻璃珠,加10 mL混合酸(4.9),加盖浸泡过夜,加一小漏斗于电炉上消解,若变棕黑色,再加混合酸,直至冒白烟,消化液呈无色透明或略带黄色,放冷,用滴管将试样消化液洗入或过滤入(视消化后试样的盐分而定)10 mL〜25 mL容量瓶中,用水少量多次洗涤锥形瓶或高脚烧杯,洗液合并于容量瓶中并定容至刻度,混匀备用;同时作试剂空白。

附录Ⅸ B 铅、镉、砷、汞、铜测定法(一部)一、原子吸收分光光度法本法系采用原子吸收分光光度法(附录Ⅴ D)测定中药材中的铅、镉、砷、汞、铜,除另有规定外,按下列方法测定。
1.铅的测定(石墨炉法)测定条件 参考条件:波长 283.3nm,干燥温度 100~120℃,持续 20 秒;灰化温度 400~750℃,持续 20~25 秒;原子化温度 1700~2100℃,持续 4~5秒;背景校正为氘灯或塞曼效应。
铅标准储备液的制备 精密量取铅单元素标准溶液适量,用 2%硝酸溶液稀释,制成每 1ml 含铅(Pb)1µg 的溶液,即得(0~5℃贮存)。
标准曲线的制备 分别精密量取铅标准储备液适量,用 2%硝酸溶液制成每1ml 分别含铅 0ng、5ng、20ng、40ng、60ng、80ng 的溶液。
分别精密量取 1ml,精密加含 1%磷酸二氢铵和 0.2%硝酸镁的溶液 1ml,混匀,精密吸取 20µl 注入石墨炉原子化器,测定吸光度,以吸光度为纵坐标,浓度为横坐标,绘制标准曲线。
供试品溶液的制备 A 法取供试品粗粉 0.5g,精密称定,置聚四氟乙烯消解罐内,加硝酸 3~5ml,混匀,浸泡过夜,盖好内盖,旋紧外套,置适宜的微波消解炉内,进行消解(按仪器规定的消解程序操作)。
消解完全后,取消解内罐置电热板上缓缓加热至红棕色蒸气挥尽,并继续缓缓浓缩至 2~3ml,放冷,用水转入 25ml 量瓶中,并稀释至刻度,摇匀,即得。
同法同时制备试剂空白溶液。
B 法取供试品粗粉 1g,精密称定,置凯氏烧瓶中,加硝酸 高氯酸(4∶1)混合溶液 5~10ml,混匀,瓶口加一小漏斗,浸泡过夜。
置电热板上加热消解,保持微沸,若变棕黑色,再加硝酸 高氯酸(4∶1)混合溶液适量,持续加热至溶液澄明后升高温度,继续加热至冒浓烟,直至白烟散尽,消解液呈无色透明或略带黄色,放冷,转入 50ml 量瓶中,用 2%硝酸溶液洗涤容器,洗液合并于量瓶中,并稀释至刻度,摇匀,即得。
1 实验部分1.1 仪器与试剂Freezer/Mill 6775液氮破碎机(SPEX SamplePrep);移液枪(BRAND);Multiwave GO 微波消解仪(Anton Paar); Agilent 5100等离子体发射光谱仪(美国Agilent);ME204E/02电子分析天平(瑞士METTLER TOLEDO)。
玻璃仪器:在1∶1硝酸溶液中浸泡24h ;微波消解罐:在5%硝酸溶液中浸泡24h 。
并用去离子水冲洗、晾干,备用。
铅、砷、铬、镉单元素标准溶液:浓度为1000μg/mL(国家标准物质中心);硝酸:优级纯;双氧水:优级纯;氢氟酸:优级纯;超纯水:18.2MΩ·cm 。
1.2 实验过程将非金属制品样块放入液氮破碎机并冷冻破碎后,准确称取0.1000g 的样品于聚四氟乙烯-四氟乙烯消解罐内,依次加入5mL 硝酸、1mL 双氧水及1mL 氢氟酸,放置在电加热板上,130℃预消解1h 后,按照表1的微波消解程序进行消解。
待整个消解完成,消解罐温度降至室温,再将罐中的溶液倒入50mL 容量瓶中,用超纯水定容并混匀,备用。
用同样的方法在不加样品的情况下制备一个空白溶液。
对澄清溶液进行测定。
表1 微波消解程序2101601531021030采用ICP-OES 光谱仪测试样品。
实验选择无其他元素明显干扰的谱线作为各元素的分析线。
铅、砷、铬、镉元素的测定谱线波长分别为220.353nm 、188.980nm 、206.502nm 及214.439nm 。
0 引言矿用非金属制品如矿用阻燃输送带、电缆、管材、风筒、电缆挂钩等因其特有的质轻、价廉、耐热性等优点,目前在矿山或矿山井下被大量使用。
由于非金属制品在生产过程中会引入大量的重金属类有害物质,在不同程度上会危害人们的健康并污染环境,这种状况已越来越引起人们的重视。
故需要对这些产品中的重金属元素的含量进行管控。
矿用非金属制品的主体材料主要为聚氯乙烯(PVC)、聚乙烯(PE)、聚丙烯(PP)、环氧树脂(EP)、三元乙丙橡胶(EPDM)及氯化聚乙烯(CPE)等,矿用非金属制品种类繁多,结构复杂,或包含上述两种以上的聚合物。
肥料中砷、镉、铬、铅、汞含量的测定肥料是农业生产中不可或缺的重要物质,它能够提供植物所需的养分,促进作物生长和增产。
然而,肥料中可能含有一些有害物质,如砷、镉、铬、铅和汞等重金属元素,它们对植物生长和人体健康都具有潜在的危害。
因此,对肥料中这些有害物质的含量进行准确测定至关重要。
砷是一种常见的有害重金属元素,它在肥料中的含量可以通过多种方法进行测定。
其中,常用的方法有原子吸收光谱法(AAS)、电感耦合等离子体发射光谱法(ICP-OES)和质谱法等。
这些方法可以快速准确地测定肥料中砷的含量,帮助农民选择合适的肥料,避免砷对作物和土壤的污染。
镉是另一种常见的有害重金属元素,它对植物生长的抑制作用尤为显著。
因此,准确测定肥料中镉的含量对于保护土壤和作物健康至关重要。
常用的测定方法有原子荧光光谱法(AFS)、电感耦合等离子体质谱法(ICP-MS)和原子吸收光谱法(AAS)等。
这些方法能够快速、准确地测定肥料中镉的含量,为农民提供合适的肥料选择建议。
铬是一种对植物生长有一定影响的重金属元素,其在肥料中的含量也需要进行准确测定。
常用的测定方法有原子吸收光谱法(AAS)、电感耦合等离子体发射光谱法(ICP-OES)和荧光光谱法等。
这些方法能够快速、准确地测定肥料中铬的含量,帮助农民选择合适的肥料,保护土壤和作物的健康。
铅是一种对人体健康具有潜在危害的重金属元素,其在肥料中的含量需要严格控制。
常用的测定方法有原子吸收光谱法(AAS)、电感耦合等离子体发射光谱法(ICP-OES)和电感耦合等离子体质谱法(ICP-MS)等。
这些方法能够准确测定肥料中铅的含量,保护农民和消费者的健康。
汞是一种极具毒性的重金属元素,其在肥料中的含量必须被严格控制。
常用的测定方法有原子吸收光谱法(AAS)、电感耦合等离子体发射光谱法(ICP-OES)和原子荧光光谱法(AFS)等。
这些方法可以快速准确地测定肥料中汞的含量,保护农民和消费者的健康。
土壤中砷铅人体可给性测试方法砷和铅是土壤中常见的重金属污染物,它们对人体健康具有重要危害。
土壤中的砷和铅可以通过吸附或富集在作物中,进而进入人体。
因此,进行土壤中砷、铅人体可给性测试非常重要,以评估土壤中重金属的潜在风险。
下面将介绍几种常用的土壤中砷、铅人体可给性测试方法。
1.直接摄入法这是一种常用的测试方法,主要通过将土壤与人体模型(如胃肠道模型)接触,模拟人体吸附土壤中砷、铅的过程。
测试中,会将经过预处理的土壤与模型溶液进行混合和搅拌,然后通过过滤、离心等操作,分离出土壤中的砷、铅。
然后,通过测定溶液中的砷、铅含量,计算出土壤中砷、铅的可溶性和人体可摄入量。
2.生物可达性法这种方法是通过使用模拟人体胃肠道的消化液处理土壤样品,并利用胆固醇(类胆汁酸)进行胃酸和肠道液的模拟,来评估土壤中砷、铅的生物可达性。
测试过程中,土壤样品与模拟胃肠液进行混合,然后通过离心、过滤等步骤,分离出溶液中的砷、铅。
最后,通过质谱仪等分析仪器对溶液中的砷、铅含量进行测定。
3.食品模型法这种方法是通过将土壤样品与常见的食品(如蔬菜、谷物等)进行混合,模拟人体通过食物摄入土壤中的砷、铅的过程。
测试中,将土壤样品与食品进行混合,并进行一定的处理(如溶解、加热等),然后提取出食品中的砷、铅。
最后,通过光谱、质谱等仪器对提取液中的砷、铅含量进行测定。
这些方法在不同的研究和监测中都有广泛应用,但需要注意,测试结果的准确性受到多种因素的影响。
首先,土壤中砷、铅的形态和特性会影响测试结果的可靠性。
其次,实验条件的选择也会对测试结果产生影响。
另外,测试结果还需要与环境标准和健康风险评估相结合,来评估土壤中砷、铅对人体的潜在风险。
总而言之,土壤中砷、铅人体可给性测试是评估土壤中重金属污染风险的重要手段,通过选择合适的测试方法和条件,可以得到可靠的测试结果,并为相应的风险评估提供重要的科学依据。
金属种铅镉砷的测定金属种铅、镉和砷的测定是一项重要的实验工作,它在环境保护、食品安全和人体健康等领域具有重要意义。
本文将介绍一种常用的测定方法,以及其在实际应用中的意义。
一、测定方法为了准确测定铅、镉和砷的含量,科学家们开发了各种分析技术。
其中,常用的方法包括原子吸收光谱法、电感耦合等离子体质谱法和火焰原子吸收光谱法等。
这些方法在测定不同样品中的金属含量时,具有高灵敏度、高准确度和高选择性的优点。
二、应用领域1. 环境保护:铅、镉和砷是环境中常见的污染物,它们对土壤、水体和大气的污染会对生态系统和人类健康造成严重影响。
通过测定它们的含量,可以及时采取有效的环境治理措施,保护生态环境的可持续发展。
2. 食品安全:铅、镉和砷是食品中常见的重金属污染物,长期摄入过量会对人体健康产生潜在风险。
通过对食品中金属含量的测定,可以及时发现和控制食品安全风险,保障人民的饮食安全。
3. 药物研发:在药物研发过程中,需要对药物中的金属杂质进行测定,以确保药物的纯度和质量。
铅、镉和砷等重金属杂质的存在可能会对药物的安全性和疗效产生不利影响,因此对其进行测定是非常重要的。
三、实验过程在进行金属种铅、镉和砷的测定实验时,首先需要收集样品,如土壤、水样、食品等。
然后,通过适当的前处理方法,将样品中的金属离子转化为易于测定的形式。
接下来,使用合适的分析方法对样品进行测定,得到各金属的含量。
最后,根据测定结果进行数据处理和分析,得出准确的测定值。
四、实际意义金属种铅、镉和砷的测定在环境和食品安全监测中具有重要应用价值。
通过测定金属含量,可以及时发现和控制污染源,保护环境和人类健康。
此外,测定金属含量还可以指导农业生产和食品加工,提高食品质量和安全性。
同时,对药物中金属杂质的测定,有助于保证药物的安全和疗效。
金属种铅、镉和砷的测定是一项具有重要意义的实验工作。
通过合适的测定方法和实验过程,可以准确测定金属含量,为环境保护、食品安全和药物研发等领域提供重要的数据支持。
食品接触用油墨铅、汞、镉、铭、碑的测定1范围本附录规定了食品接触用油墨中铅、汞、镉、格、碎元素测定的电感耦合等离子体发射光谱测定法。
本附录适用于食品接触用油墨中铅、汞、镉、铭、碑的测定。
2原理油墨经过涂膜干燥后粉碎,通过酸消解的方法转为溶液。
将所得溶液稀释定容后,各元素经电感耦合等离子体光谱仪测定强度,用标准曲线法进行定量。
3试剂和材料除非另有说明,本方法所用试剂均为光谱纯,水为GB/T6682规定的一级水。
3.1 试剂3.1.1 硝酸(HNO3)。
3.1.2 盐酸(HCI)o3.1.3 氢氟酸(HF)o3.1.4 金元素(Au)溶液(IoOOmg/1)。
3.1.5 氧气(Ar):纯度299.99%,或液氨。
3.2 试剂配制3.2.1 硝酸溶液(2+98):量取20m1硝酸,缓慢加入980m1水中,混匀。
3.2.2 硝酸溶液(1+5):量取500m1硝酸,缓慢加入250Om1水中,混匀。
3.2.3 汞标准稳定剂(1mg/1):取Im1金元素(AU)溶液,用硝酸溶液(2+98)稀释至1000m1,用于汞标准溶液的配制。
3.3 标准品元素标准储备液(IoOomg/1或100mg/1):铅、汞、镉、铭、神,采用经国家认证并授予标准物质证书的单元素或多元素标准储备液。
3.4 标准溶液配制准确吸取适量单元素标准储备液或多元素混合标准储备液,用硝酸溶液(2+98)逐级稀释配成铅、镉、格、伸混合标准曲线溶液和汞标准系列溶液,各元素浓度可参考表K铅、镉、格、础混合标准曲线溶液配制后转移至洁净的聚乙烯瓶中保存。
汞元素需要用汞标准稳定剂单独配制标准曲线溶液,配制后转移至洁净的玻璃瓶中保存。
注:可根据仪器的灵敏度、线性范围以及样液中各元素实际含量确定标准系列溶液中该元素的浓度和范围。
标准工作溶液在20C~25C下可保存2个月。
使用前摇匀。
4仪器和设备注:所有玻璃器皿均需硝酸溶液(1+5)浸泡过夜,用水反复冲洗干净。
4.1 电感耦合等离子体发射光谱仪(ICP-OES)。
美白化妆品中汞、砷、铅的测定1、汞的测定: 冷原子吸收法1.方法提要汞蒸气对波长253.7nm 的紫外光具特征吸收。
在一定的浓度范围内,吸收值与汞蒸气浓度成正比。
样品经消解、还原处理,将化合态的汞转化为原子态汞,再以载气带入测汞仪测定吸收值,与标准系列比较定量。
本方法对汞的检出限和定量下限分别为0.01 μg 和0.04 μg ,若取1g 样品测定,检出浓度为 0.01μg/g,最低定量浓度为0.04 μg/g。
2 试剂2.1 硝酸(ρ20=1.42g/mL ),优级纯。
2.2 硫酸(ρ20=1.84g/mL ),优级纯。
2.3 盐酸(ρ20=1.19g/mL ),优级纯。
2.4 过氧化氢[ω(H2 O2 )=30%]。
2.5 五氧化二钒。
2.6 硫酸[(H2 SO4 )=10%]:取硫酸(2.2)10mL,缓慢加入到90mL水中,混匀。
2.7 盐酸羟胺溶液(120g/L ):取盐酸羟胺 12.0g和氯化钠12.0g溶于100mL 水中。
2.8 氯化亚锡溶液(200g/L ):称取氯化亚锡 20g 置于250mL 烧杯中,加入盐酸(2.3)20mL,必要时可略加热促溶,全部溶解后,加水稀释至100mL。
2.9 重铬酸钾溶液(100g/L ):称取重铬酸钾 10g,溶于100mL 水中。
2.10 重铬酸钾-硝酸溶液:取重铬酸钾溶液(2.9)5mL ,加入硝酸(2.1)50mL,用水稀释至1L。
2.11 辛醇。
2.12 汞标准溶液2.12.1汞标准溶液[ ρ(Hg)=100mg/L]:称取氯化汞(HgCl2 )0.1354g 置于100mL 烧杯中,加入重铬酸钾-硝酸溶液(2.10 )溶解。
移入1000mL 容量瓶,用重铬酸钾-硝酸溶液(2.10 )稀释至刻度。
2.12.2汞标准溶液[ ρ(Hg)=10mg/L]:取汞标准溶液(2.12.1 )10.0mL 置于100mL 容量瓶中,用重铬酸钾-硝酸溶液(2.10 )稀释至刻度。
氢化物原子荧光光度法(测砷含量)
1 原理
食品试样经湿消解或干灰化后,加入硫脲使价砷预还原为三价砷,再加入硼氢化钠或硼氢化钾使还原生成砷化氢,由氩气载入石英原子化器中分解为原子态砷,在特制砷空心阴极灯的发射光激发下产生原子荧光,其荧光强度在固定条件下与被测液中的砷浓度成正比,与标准系列比较定量.
2 试剂
2.1 硼氢化钠溶液(10g/L):称取硼氢化钠2g,溶于5g/L氢氧化钾溶液200mL中,混匀(现配现用),注:此液于冰箱可保存10天,取出后应当日使用
2.2硫脲溶液(100g/L)
2.3 5%盐酸(优级纯)
2.4 砷标准使用液:移取砷标准储备液(1mg/kg)按照10倍的体积关系(最大不可超过100倍)逐级稀释至As=10ug/L
注: 砷标准溶液需用5%盐酸(优级纯)作为定容介质储存,且在定容前加入1%硫脲.
2.5硝酸—高氯酸混合液(4+1)(优级纯)
2.5 湿消解试剂:硝酸、硫酸、高氯酸(优级纯)
2.6 干灰化试剂:六水硝酸镁(150g/L)、氯化镁、盐酸(1+1)
3仪器
原子荧光光度计
4 分析步骤
4.1 试样消解
4.1.1 湿消解:称取0.5g~1.0g试样(精确至小数点后第二位),置于50~100mL 锥形瓶中,同时做试剂空白。
加10~20mL混酸,硫酸1~2mL,摇匀后放置过夜,置于电热板上加热消解。
若消解液处理至5mL左右仍有未分解物质或色泽变深,取下放冷,补加硝酸5~10mL,再消解至5mL左右观察,如此反复两三次,注意避免炭化。
如仍不能消解完全,则加入高氯酸1~2mL,继续加热消解完全后,再持续蒸发至高氯酸的白烟散尽,硫酸的白烟开始冒出。
冷却,加水20mL左右,再蒸发至冒硫酸白烟。
冷却,用水将内容物转入25mL/50mL容量瓶或比色管中,加入硫脲溶液(100g/L)2.5mL,补水至刻度并混匀,放置30min后备测
4.1.2 干灰化:一般应用于固体试样。
称取0.5~1.0g(精确至小数点后第二位)于50mL坩埚中,同时做试剂空白。
加入六水硝酸镁(150g/L)10mL混匀,低热蒸干,将氧化镁1g仔细覆盖在干渣上,于电炉上炭化至无黑烟,移入550℃高温电炉灰化4h,取出放冷,小心加入(1+1)盐酸10mL以中和氧化镁并溶解灰分,转入25mL/50mL容量瓶或比色管中,加入硫脲溶液(100g/L)2.5mL/
5.0mL,补酸至刻度并混匀,放置30min后备测
5 样品测定
5.1 按照原子荧光操作规程测定样品
5.2 载流液; 5%盐酸
5.3 仪器参数:仪器本身默认值
6 结果计算
仪器自动计算结果
7 精密度
湿消解法在重复性条件下获得的两次独立测定结果的绝对差值不得超过算术平均值的10%
干灰化法在重复性条件下获得的两次独立测定结果的绝对差值不得超过算术平均值的15%
氢化物原子荧光光度法(测铅含量)
1 原理
食品试样经酸热消化后,在酸性介质中,试样中的铅与硼氢化钠或硼氢化钾反应生成挥发性铅的氢化物。
由氩气载入石英原子化器中原子化,在特制铅空心阴极灯的发射光激发下,基态铅原子被激发至高能态;在去活化回到基态时,发射出特征波长的荧光,其荧光强度与铅含量成正比,根据标准系列进行定量。
2 试剂
2.1 硼氢化钾溶液(10g/L):称取硼氢化钾2g,铁氰化钾2g,溶于10g/L氢氧化钾溶液200mL中,混匀(现配现用),注:此液于冰箱可保存10天,取出后应当日使用
2.2草酸溶液(10g/L):称取1.0g草酸,加入溶解至100mL,混匀
2.3 1%硝酸(优级纯)
2.4铅标准使用液:移取砷标准储备液(1mg/kg)按照10倍的体积关系(最大不可超过100倍)逐级稀释至Pb=10ug/L
注: 铅标准溶液需用1%硝酸(优级纯)作为定容介质储存,且在定容前加入0.5mL草酸
2.5硝酸—高氯酸混合液(4+1)(优级纯)
3仪器
原子荧光光度计
4 分析步骤
4.1 试样消解
4.1.1 湿消解:称取0.5g~1.0g试样(精确至小数点后第二位),置于50~100mL 锥形瓶中,同时做试剂空白。
加10~20mL混酸,摇匀后放置过夜,置于电热板上加热消解,至消化液呈淡黄色或无色(如消解过程中色泽较深,稍冷补加少量硝酸,继续消解),稍冷加入20mL水再继续加热赶酸,至消解液烧干(有盐类物质析出),放冷,用水将其转入25mL容量瓶或比色管中,加入0.5mL 草酸,补水至刻度并混匀,放置30min后备测
5 样品测定
5.1 按照原子荧光操作规程测定样品
5.2 载流液; 1%硝酸
5.3 仪器参数:仪器本身默认值
6 结果计算
仪器自动计算结果
7 精密度
湿消解法在重复性条件下获得的两次独立测定结果的绝对差值不得超过算术平均值的10%。