数字基因表达谱
- 格式:pdf
- 大小:944.18 KB
- 文档页数:1

作物学报 ACTA AGRONOMICA SINICA 2013, 39(6): 1045−1053/zwxb/ ISSN 0496-3490; CODEN TSHPA9E-mail: xbzw@本研究由国家现代农业产业技术体系建设专项(CARS-14-07B), 国家自然科学基金项目(31101177)和山东省自然科学基金项目(ZR2011CQ027)资助。
*通讯作者(Corresponding author): 万勇善, E-mail: yswan@, Tel: +86 (0)538 8241540第一作者联系方式: E-mail: saqsshh@ **同等贡献(Contributed equally to this work)Received(收稿日期): 2012-07-06; Accepted(接受日期): 2013-01-15; Published online(网络出版日期): 2013-02-19. URL: /kcms/detail/11.1809.S.20130219.1020.007.htmlDOI: 10.3724/SP.J.1006.2013.01045花生干旱胁迫响应基因的数字表达谱分析孙爱清1,** 张杰道2,** 万勇善1,* 刘风珍1 张 昆1 孙 利11山东农业大学农学院 / 作物生物学国家重点实验室 / 山东省作物生物学重点实验室, 山东泰安 271018; 2山东农业大学生命科学学院, 山东泰安 271018摘 要: 以抗旱性强的花生品种丰花5号为材料, 利用Solexa 高通量测序技术对15% PEG 处理后的花生叶片cDNA 文库进行差异基因表达谱分析。
结果表明, 转录组基因表达表现出高度的不均一性和冗余性, 低于10个拷贝的标签占总标签种类的73.1%, 但其表达量只占总标签表达量的9.0%。
根据已知序列信息鉴定出935个差异表达基因, 其中64.5%下调表达。

整合数字基因表达谱与全基因组关联分析鉴定猪血液性状候选基因徐盼;张震;章峰;杨斌;段艳宇【期刊名称】《中国农业科学》【年(卷),期】2016(000)002【摘要】【目的】整合数字基因表达谱与全基因组关联分析鉴定白色杜洛克×二花脸 F2资源群体的血液性状候选基因。
【方法】白色杜洛克×二花脸F2资源群体在(240±3)d屠宰,收集血液于抗凝管中进行血常规检测。
利用Illumina 60K SNP芯片对1020头F2资源群体进行基因分型。
剔除基因型检出率<90%和孟德尔错误检出率>5%的个体。
检出率<95%、次等位基因频率<5%、哈代-温伯格检验(HWE)P <5×10-6、与性染色体连锁疑似常染色体的SNP被筛除。
利用Illumina GA II 测序仪测序对502头F2资源群体的肝脏进行数字基因表达谱测序。
测序得到的原始数据经过滤获得清洁标签后与参考标签数据库比对,将能唯一比对到参考基因序列的清洁标签数量进行标准化处理以获得标准化的基因表达量。
每个转录本的表达水平进一步转化为 lg2值。
在少于20%的个体中表达的转录本被滤去。
表型性状和基因表达性状使用R程序包中GenABEL内polygentic功能进行性别、批次和亲缘关系的校正。
其残差使用R程序包中斯皮尔曼系数评估基因表达水平与表型数据的关联性,设定保守阈值P<5×10-4时调整多重检验。
将检测到的表达数量性状位点(eQTL)及其对应基因根据其位置相对照的关系进行绘图。
搜寻前期GWAS最高点5.0 Mb区域内eQTL结合GWAS结果进行综合分析。
Gene Ontology & KEGG pathway富集分析使用在线工具DAVID。
基因共表达网络使用在线工具GeneMANIA进行构建。
【结果】白色杜洛克×二花脸F2资源群体中502个个体的20108个肝脏转录本通过了质检。

基因表达谱分析在药物研究中的应用在众多的药物研究中,基因表达谱分析已经逐渐成为一种能够有效提高药物研究效率的工具。
作为一种新颖的基因组学技术,它可以快速地分析人体内基因的表达情况,并识别与特定疾病相关的基因。
这种技术已经在许多的药物研究中成功应用,这里将会具体介绍基因表达谱分析在药物研究中的应用。
基因表达谱分析简介基因表达谱分析是一种可以追踪特定基因在特定条件下的转录活动量的方法。
该方法结合基因组学、生物信息学、计算机科学和生物学于一体,可以为研究人员提供一系列有关基因表达的数据,包括基因转录过程中产生的mRNA量。
此外,基因表达谱分析还可以通过测量RNA分子在细胞内的存在量,从而识别细胞类型、状态以及其所在环境。
总体来说,基因表达谱分析可以为药物研究提供大量的基础信息。
基因表达谱分析的应用1.寻找新的药物靶点通过基因表达谱分析,可以了解到特定疾病患者基因的表达情况。
这让科学家们有了更深层次的认识和了解相关病理生理特征。
比如,目前就有很多疾病是由于基因表达失调导致的,比如乳腺癌、大肠癌、肝炎等。
通过基因表达谱分析,药物研究人员可以识别新的药物靶点及开发新的药物治疗方法。
2.评估药物疗效药物的疗效是影响药物研究的重要因素之一。
通过基因表达谱分析,科学家们可以获得药物与靶点蛋白相互作用所涉及到的相关基因信息,这样就能对药物的疗效做出更加准确的评估。
比如,科学家们发现使用某种特定 Compound A治疗非小细胞肺癌患者可以降低基因P13K/AKT/mTOR的表达量,这些表达量的下降是由Compound A对癌症细胞中PI3K/Akt/mTOR的电荷阻断所引起的,可以更好的评估Compound A的疗效。
3.抗药性研究许多患者在使用药物治疗之后会形成抗药性,这是药物研究人员在后续工作中需关注和解决的一个问题。
通过基因表达谱分析,可以发现在基因层面上抗药性基因的表达量增多。
这些基因是抗药性形成的重要因素,对其进行研究并找出相应药物突破可以使药物研究有所突破。

转录组RNAseq术语解释RNA-Seq名词解释1.inde某2.碱基质量值(QualityScore或Q-core)是碱基识别(BaeCalling)出错的概率的整数映射。
碱基质量值越高表明碱基识别越可靠,碱基测错的可能性越小。
3.Q30碱基质量值为Q30代表碱基的精确度在99.9%。
4.FPKM (FragmentPerKilobaeoftrancriptperMillionfragmentmapped)每1百万个map上的read中map到外显子的每1K个碱基上的fragment个数。
计算公式为公式中,cDNAFragment表示比对到某一转录本上的片段数目,即双端Read数目;MappedRead(Million)表示MappedRead总数,以10为单位;TrancriptLength(kb):转录本长度,以kb个碱基为单位。
5.FC(FoldChange)即差异表达倍数。
6.FDR(FaleDicoveryRate)即错误发现率,定义为在多重假设检验过程中,错误拒绝(拒绝真的原(零)假设)的个数占所有被拒绝的原假设个数的比例的期望值。
通过控制FDR来决定P值的阈值。
7.P值(P-value)即概率,反映某一事件发生的可能性大小。
统计学根据显著性检验方法所得到的P值,一般以P<0.05为显著,P<0.01为非常显著,其含义是样本间的差异由抽样误差所致的概率小于0.05或0.01。
8.可变剪接(Alternativeplicing)有些基因的一个mRNA前体通过不同的剪接方式(选择不同的剪接位点)产生不同的mRNA剪接异构体,这一过程称为可变剪接(或选择性剪接,alternativeplicing)。
可变剪接是调节基因表达和产生蛋白质组多样性的重要机制,是导致真核生物基因和蛋白质数量较大差异的重要原因。
在生物体内,主要存在7种可变剪接类型:A)E某onkipping;B)Intronretention;C)Alternative5'pliceite;D)Alternative3'pliceite;E)Alternativefirte某on;F)Alternativelate某on;G)Mutuallye某cluivee某on。

表达谱数据表达谱数据是一种描述基因在特定条件下表达水平的数据,可以反映基因在生物体内的活跃程度和重要作用。
表达谱数据通常是通过高通量测序技术获得的,例如RNA-Seq或microarray技术。
这些数据对研究基因功能及其对生物过程的贡献非常重要,因为它们可以揭示基因的功能、代谢途径和信号通路等复杂的生物过程。
表达谱是生物数据科学领域的主流,它可以用于构建生物系统的基因调控网络图,从而去了解生物物种内部的调控机制,从而识别出治疗基因表达异常疾病的潜在靶点。
表达谱数据的数据类型和学科领域是多如牛毛的,涵盖了癌症、神经学、生殖生物学、免疫学和感染病理学等众多领域。
表达谱数据可以用于许多不同的目的,例如预测细胞的类型、研究细胞的生物学特性、发现新的医疗目标等。
表达谱数据的分析包括预处理、特征提取和建模等步骤。
预处理步骤包括数据过滤、归一化、批次效应调整和缺失数据填充等,这是优化数据质量的必要步骤。
特征提取步骤包括细胞类型、生物过程、调节机制和代谢途径等关键生物学特征的提取。
建模是指将表达谱数据与其他类型的数据整合,例如蛋白质互作数据或基因组测序数据。
这就是基因表达,调控网络和系统生物学等领域的研究方向。
表达谱数据的应用范围非常广泛,从基础科学到应用科学都有着重要作用。
例如,在基础研究方面,表达谱数据可以用于研究基因调控以及基因的功能,从而进一步了解遗传学机制和生物进化的过程。
在应用科学中,表达谱数据可以用于分析药物的作用和安全性,发现影响药物代谢和毒性的基因,并为药物研发提供重要的参考信息。
另外,表达谱数据也可以用于生产领域,例如利用表达谱数据优化工业培养基和工业生物发酵生产过程,提高生产效率。
在生物安全领域,表达谱数据可以用于分析致病菌基因的表达水平,从而开发针对它们的抗菌药物。
在农业领域,基于表达谱数据,可以研究农作物的病害抵抗性、调节所属基因功能等方向进行研究。
总的来说,表达谱数据是重要的生物信息学研究领域,有着广泛的应用前景和潜在的医疗价值。
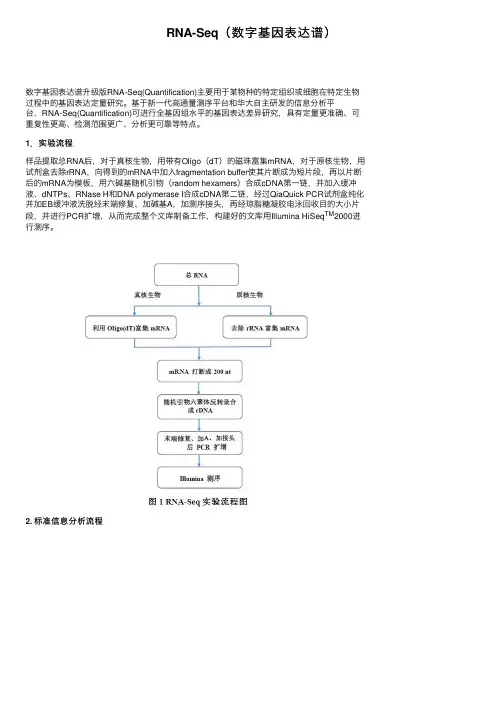
RNA-Seq(数字基因表达谱)数字基因表达谱升级版RNA-Seq(Quantification)主要⽤于某物种的特定组织或细胞在特定⽣物过程中的基因表达定量研究。
基于新⼀代⾼通量测序平台和华⼤⾃主研发的信息分析平台,RNA-Seq(Quantification)可进⾏全基因组⽔平的基因表达差异研究,具有定量更准确、可重复性更⾼、检测范围更⼴、分析更可靠等特点。
1.实验流程样品提取总RNA后,对于真核⽣物,⽤带有Oligo(dT)的磁珠富集mRNA,对于原核⽣物,⽤试剂盒去除rRNA,向得到的mRNA中加⼊fragmentation buffer使其⽚断成为短⽚段,再以⽚断后的mRNA为模板,⽤六碱基随机引物(random hexamers)合成cDNA第⼀链,并加⼊缓冲液、dNTPs、RNase H和DNA polymerase I合成cDNA第⼆链,经过QiaQuick PCR试剂盒纯化并加EB缓冲液洗脱经末端修复、加碱基A,加测序接头,再经琼脂糖凝胶电泳回收⽬的⼤⼩⽚段,并进⾏PCR扩增,从⽽完成整个⽂库制备⼯作,构建好的⽂库⽤Illumina HiSeq TM2000进⾏测序。
2. 标准信息分析流程3.技术优势⾼重复性通过对UHRR和HBRR标准品进⾏重复性研究(如图3)证明,两个样本的技术重复相关系数均可达0.99以上,可见,RNA-Seq(Quantification)具有极好的技术重复性。
检测阈值宽从图4中基因表达量检测范围看,RNA-Seq(Quantification)不仅检测范围⽐Affymetrix芯⽚宽,⽽且⽐Affymetrix更易检测到低丰度的基因。
定量准确⽤qRT-PCR和RNA-Seq(Quantification)两种⽅法进⾏UHRR和HBRR基因表达差异研究,结果相关性如图5所⽰: RNA-Seq (Quantification)与qPCR研究结果相关系数为0.915,表明RNA-Seq (Quantification)技术定量准确性⾼。

使用生物大数据中心数据库进行基因表达谱分析的步骤生物大数据中心数据库是一个强大的工具,可以用于分析基因表达谱。
在进行基因表达谱分析之前,我们需要明确几个步骤。
本文将详细介绍如何使用生物大数据中心数据库进行基因表达谱分析。
第一步是向生物大数据中心数据库注册账号并登录。
注册账号是使用生物大数据中心数据库进行基因表达谱分析的第一步。
可以访问该数据库的官方网站进行注册。
填写个人信息、用户名和密码后,您将获得一个账号。
登录之后,您可以访问数据库的各个功能和工具。
第二步是选择合适的基因表达数据集。
生物大数据中心数据库拥有众多的基因表达数据集,您可以根据自己的研究需求选择合适的数据集。
数据集通常被分类为不同的物种、组织类型和疾病状态。
例如,如果您的研究关注人类心脏组织的基因表达谱,您可以选择包含心脏组织样本的数据集。
第三步是导入和预处理基因表达数据。
一旦选择了适当的数据集,您可以根据需要下载数据集中的原始数据。
原始数据通常以文本文件或Excel文件的形式提供。
在导入数据之前,您可能需要进行一些预处理步骤,例如去除噪声、归一化或筛选不感兴趣的基因。
这些预处理步骤可以使用生物大数据中心数据库中的工具完成。
第四步是进行基因表达谱分析。
生物大数据中心数据库提供了各种分析工具,可以帮助您更好地理解基因表达谱。
其中包括差异表达基因分析、基因共表达网络分析、功能富集分析等。
差异表达基因分析可以帮助您识别在不同样本之间表达水平显著不同的基因。
基因共表达网络分析可以帮助您发现在相似组织或条件下共同表达的基因模块。
功能富集分析可以帮助您理解哪些生物学过程和信号通路参与了基因的调控。
这些工具可以根据您的研究需求进行灵活的组合和调整。
第五步是解释和呈现分析结果。
一旦完成了基因表达谱分析,您将得到大量的结果,包括差异表达基因列表、共表达基因模块和功能富集结果。
解释和呈现这些结果对于得到有意义的结论至关重要。
生物大数据中心数据库通常提供了数据可视化和分析结果导出的功能。

基因组学研究中的表达谱数据分析方法解析概述:基因组学研究是研究生物体基因组的编码和非编码序列的科学。
在基因组学研究中,表达谱数据是一种重要的数据类型,由于其高维度和复杂性,需要采用一系列的分析方法和技术来解析。
本文将介绍基因组表达谱数据的分析方法,包括数据预处理、差异表达分析、聚类分析、富集分析以及网络分析。
一、数据预处理:数据预处理是基因组表达谱数据分析的第一步,目的是清除原始数据中的噪声、去除非生物学的变异以及纠正技术上的偏见。
常用的数据预处理步骤包括数据质量控制、归一化和基因过滤。
1. 数据质量控制:首先需要对原始数据进行质量控制,该步骤可通过查看测序质量分数和测序错误率来评估。
常用的工具有FastQC和Trimmomatic等。
该步骤的目的是排除测序引入的噪声。
2. 归一化:由于不同样本之间的表达量存在显著的差异,我们需要对数据进行归一化处理,以消除样本间的偏差。
常用的归一化方法有TPM、FPKM和RPKM等。
归一化后的数据便于后续的比较和统计分析。
3. 基因过滤:在分析表达谱数据时,一些基因的表达量非常低,对分析结果产生较小的影响并增加运算复杂性。
因此,我们通常会对表达量低于一定阈值的基因进行过滤处理,从而提高分析效率。
常用的过滤标准包括表达量百分位数和表达量阈值。
二、差异表达分析:差异表达分析是基因表达谱数据分析的核心内容之一,旨在发现不同条件下存在差异表达的基因。
通常,差异表达分析包括基于假设检验的方法和机器学习方法。
1. 基于假设检验的方法:这类方法通常基于统计学原理,将样本分组,通过计算差异表达的显著性水平来判断基因是否差异表达。
常用的方法包括Student's t-test、Wilcoxon秩和检验和Fisher's确切检验等。
这些方法基于不同的假设,在数据有明确的分布前提下,可以得到比较可靠的差异表达结果。
2. 机器学习方法:机器学习方法对差异表达分析具有较高的灵活性和预测能力。

基因表达谱的构建和分析方法基因表达谱是指某一时刻细胞内基因转录水平的全面反映。
它对了解不同细胞状态的差异性、疾病发生机制及药物治疗等具有重要的意义。
本文将对基因表达谱的构建和分析方法进行简要介绍。
一、基因表达谱的构建基因表达谱的构建方法包括microarray和RNA-Seq两种主要技术方法。
1. microarraymicroarray技术是将探针(probe)固定在芯片表面用于检测不同的核酸分子。
其构建基因表达谱的流程如下:(1)提取全基因组mRNA,反转录为cDNA。
(2)将cDNA打标记并杂交到微阵列中。
(3)信号扫描与数据分析。
microarray技术具有高通量、快速、灵敏、重复性好等特点,被广泛应用于药物筛选、肿瘤检测和疾病诊断等领域。
但是,其局限在于存在信号的非特异性、探针设计的错误等问题。
2. RNA-SeqRNA-Seq技术是基于高通量测序技术,通过定量并分析RNA 样本中所有的转录本、可变剪切事件和基因表达状况。
其构建基因表达谱的流程如下:(1)提取RNA,并用RNA脱除重复序列技术去除rRNA。
(2)转录为cDNA。
(3)建立文库并测序。
(4)数据处理和分析。
RNA-Seq技术具有更高的分辨率和准确度,能够检测到新转录本和SNP,且不受局限于预先设定的探针。
但其存在成本、数据处理和分析的复杂度等问题。
二、基因表达谱的分析方法基因表达谱的分析方法包括聚类分析、差异表达基因分析、通路富集分析等多种方法,这里仅简要介绍其中的两种。
1. 聚类分析聚类分析可以将一组基因根据其表达特征分成不同的簇,并确定它们之间的相似度。
聚类分析是基于特征基因进行的,特征基因的数量对结果有重要影响。
聚类分析主要分为两种:层次聚类和k-means聚类。
层次聚类根据相似度建立基因树,然后根据阈值将基因分为不同的簇。
k-means聚类将基因分成固定数量的簇,通过相似度计算和簇内距离最小化来划分簇。
2. 差异表达基因分析差异表达基因分析用于比较两个或多个条件下基因表达水平的差异。

基因表达谱分析技术的原理与方法随着基因组学技术的发展,我们可以从一个细胞或组织中同时检测数以万计的基因,了解人体健康和病理的分子机制。
基因表达谱分析技术,又称转录组学技术,是一种重要的基因组学技术,它可以帮助我们深入了解基因表达的变化及其对生物学特征和疾病的影响。
在本篇文章中,我们将介绍基因表达谱分析技术的原理和常用方法。
原理基因编码不同功能蛋白的RNA是由基因的转录过程产生。
基因表达是指在特定的时间点和组织中转录某一基因所产生的RNA数量和质量。
例如,心脏细胞和肝脏细胞表达不同的基因,因为它们需要不同的蛋白质来执行其特定功能。
基因表达谱分析技术就是通过检测RNA水平的变化来揭示不同组织、疾病和情况下基因的表达变化。
在基因表达谱分析中,采集组织或细胞的RNA,把RNA转化为cDNA,再将cDNA探针的引物或/和微阵列片段引入cDNA上进行探针测序或比较。
探针把其考察的基因特异性的cDNA附着在cDNA探针上,然后将其组分检测出。
在反转录,多聚酶链反应(PCR)或减少串接的基础上,引物是特异探针或一段数字长cDNA中的一个段落,被称作探针序列,以检测在RNA大样本中是否有包含这样的特异性片段。
通过这种方法,我们可以得到不同组织或情况下的RNA表达状况,从而分析基因表达谱。
方法1.微阵列微阵列是最常用的基因表达谱分析技术之一。
在微阵列上,数千个cDNA探针被绑定到玻璃片上,每个探针用来检测一个特定的基因。
将RNA转化成标记染料的cDNA,将其添加到微阵列上,并运用一些特殊的分子技术比如荧光检测或电化学检测等,检测cDNA与微阵列上的探针结合的信号。
这种方法非常适合于同时分析数千个基因,在研究基因调控网络及其调节中扮演重要角色时,微阵列可以很好地对大规模基因表达谱的分析。
2. RNA测序RNA测序技术已成为转录组分析领域的领导者。
它可以直接检测RNA而不需要提前知道基因序列,而且这种技术不受在微阵列上的探针长度或性能的影响。
Small RNA:生物体内一类高度保守的重要的功能分子,其大小在18-30nt,包括microRNA、siRNA、snRNA、snoRNA和piRNA(piwi-interacting RNA)等,它的主要功能是诱导基因沉默,调控细胞生长、发育、基因转录和翻译等生物学过程。
以miRNA为例介绍它们的功能:miRNA与RNA诱导沉默复合体(RNA induced silencing complex, RISC)结合,并将此复合体与其互补的mRNA序列结合,根据靶序列与miRNA的互补程度,从而导致靶序列降解或干扰靶序列蛋白质的翻译过程。
SD 区域:Segment duplication,串联重复是由序列相近的一些DNA 片段串联组成。
串联重复在人类基因多样性的灵长类基因中发挥重要作用。
Genotype and phenotype:基因型与表型,基因型是指某一生物个体全部基因组合的总称;表型,又称性状,是基因型和环境共同作用的结果。
基因组:Genome,单倍体细胞核、细胞器(线粒体、叶绿体)或病毒粒子所含的全部DNA 分子或RNA分子。
全基因组de novo测序:又称从头测序,它不依赖于任何现有的序列资料,而直接对某个物种的基因组进行测序,然后利用生物信息学分析手段对序列进行拼接、组装,从而获得该物种的基因组序列图谱。
全基因组重测序:对已有参考序列(Reference Sequence)物种的不同个体进行基因组测序,并以此为基础进行个体或群体水平的遗传差异性分析。
全基因组重测序能够发现大量的单核苷酸多态性位点(SNP)、拷贝数变异(Copy Number Variation,CNV)、插入缺失(InDel,Insertion/Deletion)、结构变异(Structure Variation,SV)等变异类型,以准确快速的方法将单个参考基因组信息上升为群体遗传特征。
转录组:Transcriptome,是指特定生长阶段某组织或细胞内所有转录产物的集合;狭义上指所有mRNA的集合。
Small RNA:生物体一类高度保守的重要的功能分子,其大小在18-30nt,包括microRNA、siRNA、snRNA、snoRNA和piRNA(piwi-interacting RNA)等,它的主要功能是诱导基因沉默,调控细胞生长、发育、基因转录和翻译等生物学过程。
以miRNA为例介绍它们的功能:miRNA与RNA诱导沉默复合体(RNA induced silencing complex, RISC)结合,并将此复合体与其互补的mRNA序列结合,根据靶序列与miRNA的互补程度,从而导致靶序列降解或干扰靶序列蛋白质的翻译过程。
SD 区域:Segment duplication,串联重复是由序列相近的一些DNA 片段串联组成。
串联重复在人类基因多样性的灵长类基因中发挥重要作用。
Genotype and phenotype:基因型与表型,基因型是指某一生物个体全部基因组合的总称;表型,又称性状,是基因型和环境共同作用的结果。
基因组:Genome,单倍体细胞核、细胞器(线粒体、叶绿体)或病毒粒子所含的全部DNA 分子或RNA分子。
全基因组de novo测序:又称从头测序,它不依赖于任何现有的序列资料,而直接对某个物种的基因组进行测序,然后利用生物信息学分析手段对序列进行拼接、组装,从而获得该物种的基因组序列图谱。
全基因组重测序:对已有参考序列(Reference Sequence)物种的不同个体进行基因组测序,并以此为基础进行个体或群体水平的遗传差异性分析。
全基因组重测序能够发现大量的单核苷酸多态性位点(SNP)、拷贝数变异(Copy Number Variation,CNV)、插入缺失(InDel,Insertion/Deletion)、结构变异(Structure Variation,SV)等变异类型,以准确快速的方法将单个参考基因组信息上升为群体遗传特征。
转录组:Transcriptome,是指特定生长阶段某组织或细胞所有转录产物的集合;狭义上指所有mRNA的集合。
基因表达谱的分析和解读基因表达谱是指生物体内基因在特定环境或状态下的表达情况的记录,是基因组学、分子生物学和计算生物学的交叉学科。
目前,随着高通量测序技术和计算能力的迅猛发展,基因表达谱分析逐渐成为生命科学研究的重要领域。
一、基因表达谱的分析1、测定基因表达谱基因表达谱的测定主要有两种方法:芯片技术和转录组测序。
芯片技术是通过制备特定的DNA探针,然后将其固定到芯片表面,用于检测样品中的RNA,可以同时检测几百万个基因。
转录组测序则是通过高通量测序技术,对RNA进行测序,可以获取到全基因组的表达信息。
两种方法具有互补性,可以提供更为全面的基因表达谱信息。
2、处理基因表达谱数据分析基因表达谱数据的主要任务是将大量的原始数据转化为可解释和可视化的结果。
常用的数据处理方法包括以下几个步骤:(1)数据归一化:由于样品之间的RNA浓度和RNA种类的差异,需要进行数据归一化,以消除这些技术差异。
(2)差异分析:根据生物实验的目的,选择适宜的分析方法,比较不同样品在基因表达水平上的差异。
(3)聚类分析:聚类分析可以将相似的基因表达谱分为一组,便于发掘潜在的基因功能和作用途径。
二、基因表达谱的解读1、生物信息学分析基因表达谱数据的解析和生物信息学密切相关。
常见的生物信息学分析包括基因富集分析、通路富集分析和功能注释分析。
基因富集分析是通过将基因表达谱中显著性差异的基因与特定的基因功能数据库相比较,来鉴定具有显著富集的通路和生物过程。
通路富集分析则是将差异基因与已知通路或生物过程相匹配,以确定哪些通路或过程与表型变化相关。
2、机器学习方法机器学习是一种人工智能的分析方法,目的是从数据中挖掘模式和规律。
基于机器学习的基因表达谱分类方法可以将样本分为不同的亚型或状态,以进一步理解基因表达谱的生物学意义。
常见的机器学习方法包括支持向量机、随机森林和人工神经网络等。
机器学习方法通常需要多个数据集的共同验证,以确保分析的稳健性和可靠性。
基因表达谱和基因功能注释的分析方法随着生物学领域的不断发展,我们对于基因表达和功能的了解也越来越深刻。
基因表达谱和功能注释是分析与解释基因信息的重要方法。
本文将介绍基因表达谱和基因功能注释的分析方法,包括技术原理、分析流程、数据处理等内容,希望能让读者对这方面的知识有更深入的认识和理解。
一、基因表达谱分析基因表达谱是衡量不同细胞、组织或条件下基因表达水平的方法。
它能够揭示基因间的关系,帮助研究人员了解生命体系中基因的功能和调控机制。
而基因表达谱分析是基于高通量技术的方法,用于鉴定和描述特定条件下基因表达的差异性。
下面我们将介绍主要的方法和技术。
1. 基因芯片技术基因芯片技术通过对芯片进行杂交实现对千万个基因进行同时分析。
这种技术被广泛应用于各种研究领域,尤其是对于基因组研究和疾病研究。
但是基因芯片的优势也伴随着一些局限性,比如感知度和特异性等缺点,所以近年来更加应用的是RNA-seq技术。
2. RNA-seq技术RNA-seq技术是通过将RNA分子转化为cDNA来进行分析的。
相比于其他方法,RNA-seq有许多优势,如高灵敏度、更广泛的检测范围和能够分析具有新的转录本,可变剪切异构体和外显子的新基因等。
但是它的分辨率和灵敏度也有一些局限性。
这两种技术都是分析基因表达谱的主流方法。
下面我们将详细的介绍分析流程。
1. 数据获取和清理首先需要从获取基因表达数据,比如在NCBI,GEO,和ArrayExpress等平台可以找到大量的数据资源。
接下来进行数据清理,将原始数据进行预处理,如数据质量控制和归一化处理等。
2. 差异分析差异分析是基因表达谱分析的主要流程,其中涉及许多基于统计的方法和算法。
通过比较不同实验条件下的基因表达差异,我们可以发现哪些基因与哪些疾病或者生理现象有关,从而研究基因调控的模式和机制。
3. 功能注释差异分析的结果就是一大堆数字,需要进行功能注释才能具备生物学意义。
基因功能注释就是为每个差异基因分配一些生物学注释,如基因本体分析,KEGG通路分析等,这样就可以更好的了解差异基因的功能模式。
基因表达谱基因表达谱是一种系统性研究,利用分子生物学技术,研究不同基因是如何表达的,从而探索基因表达规律及其对生物存在的重要影响。
它是现代生物学的基础,为解释生命现象提供了重要参考。
基因表达谱的建立是分子生物学的重要任务之一,在过去的几十年中得到了很大的突破。
基因表达谱的研究不仅揭示了基因如何调控彼此之间的表达量以及如何响应外界刺激,而且给出了每个基因表达量的精确数据,为我们理解基因表达调控过程提供了重要的研究信息。
研究基因表达谱的方法有许多,其中最常用的方法是定量PCR (qPCR)和DNA微阵列技术(简称DNA阵列技术)。
定量PCR是一种可以定量检测特定基因表达量的实验方法,它可以检测出微量的物质,并可以进行定量比较。
DNA阵列技术是一种把多个基因同时检测的技术,能够同时检测数千个基因的表达量,这种技术可以检测出多个基因表达量的变化,可以给出完整的基因表达谱。
基因表达谱的研究为生物学的发展提供了重要的支持。
它不仅给生物学的研究提供了一个重要研究视角,也为我们了解基因在生物体内的表达谱提供了有价值的定量数据。
例如,基因表达谱研究可以帮助我们了解基因及其调控机制,从而为病理生理研究以及药物筛选提供重要的信息,如抗病毒药物的研发、癌症治疗的设计等。
同时,基因表达谱的研究也极大地促进了生物计算和数据分析领域的发展。
由于基因表达谱研究得到了大量高质量的基因数据,生物信息学家可以使用这些数据进行模式建构和数据挖掘,从而帮助我们更深入地了解基因表达调控机制,以及基因如何影响细胞及机体间的各个层次。
总之,基因表达谱研究是现代生物学的一个重要组成部分,其研究成果在细胞、分子及系统生物学研究中都起着重要的作用。
未来,基因表达谱的研究仍将继续发展,并在多种领域发挥越来越重要的作用,为更好的了解生命的规律,营造人类健康的环境提供重要的支持。