转录组高通量测序转录组数据分析差异表达基因分析 PPT
- 格式:ppt
- 大小:2.15 MB
- 文档页数:67

生物信息学中转录组数据分析方法与差异表达基因鉴定近年来,随着高通量测序技术的快速发展,生物信息学成为了生物学研究不可或缺的一部分。
而转录组数据分析作为生物信息学的重要方法之一,在研究生物体内基因表达变化、差异表达基因的鉴定以及功能注释等方面发挥着重要作用。
转录组数据分析的第一步是将测序得到的原始数据进行质控与清洗。
在这个步骤中,需要运用一系列的质量评估工具来评估原始数据的质量,并且对数据中的低质量序列、接头污染等进行去除,以保证后续分析的准确性和可靠性。
之后,对于清洗后的测序数据,可以进行基因定量分析。
基因定量可以通过统计数据中每个基因的发现数目来实现,以探究不同样本中基因的表达量差异。
最常见的基因定量方法是将每个样本的测序数据比对到参考基因组,然后使用一些特定的算法来计算每个基因的读数。
最常用的算法包括RPKM(reads per kilobase million)和TPM(transcripts per million)等。
在基因定量的基础上,可以进行差异表达基因的鉴定。
差异表达分析旨在找出在不同条件下表达水平发生显著变化的基因。
常用的差异表达分析方法包括DESeq2、edgeR和limma等。
这些方法基于统计学原理,通过对基因表达进行归一化、方差稳定化和模型的拟合来找出差异表达基因。
除了差异表达基因的鉴定,还可以进行富集分析,以确定差异表达基因的功能。
富集分析是一种将某一特定基因集与已知功能注释的基因集相比较的方法,可以帮助研究人员了解差异表达基因在生物学过程和通路中的功能变化。
在这个步骤中,通常会使用一些公共数据库,如GO(Gene Ontology),KEGG(Kyoto Encyclopedia of Genes and Genomes)和Reactome等,来获得基因功能注释信息。
除了上述提到的方法,还有一些其他的转录组数据分析方法可供选择。
例如,可以使用聚类分析方法对样本进行聚类,以寻找具有类似表达模式的基因组。

基于转录组数据的差异表达基因分析转录组(transcriptome)是指一个生物在一定时间和环境下其全体细胞内的RNA转录产物的总和,即所有可以转录的基因产物,包括编码RNA和非编码RNA。
转录组分析(transcriptome analysis)是一种研究基因表达级别、结构和调控机制的手段,通过对RNA测序数据的研究,可以更好地理解生物体内基因的调控规律和生命活动的本质。
其中,差异表达基因(differentially expressed genes,DEGs)分析是转录组分析的一种重要方法,可以帮助我们研究基因的功能和作用,深入探究生物组织和功能间的关系。
一、差异表达基因的概念差异表达基因是指在相同条件下两种或多种样本中差异表达的基因,即其表达水平在不同样本中差异显著,这种差异有时也称为基因表达的变化或调控。
常用的方法是对不同样本中的转录组进行测序,获得每个基因的表达量,再通过各种差异分析方法,筛选出差异表达的基因,这些基因可能具有重要的生物学功能,可以在不同种类的研究中得到应用。
二、差异表达基因分析的步骤差异表达基因分析一般包括以下步骤:1. 数据处理:包括数据清洗、对齐和定量等步骤。
转录组测序数据从测序平台获取后需要进行数据清洗,包括去除低质量的序列、去除污染序列和引物序列等。
然后需要将原始序列数据对齐到参考基因组或类似序列上,例如转录本、参考转录本或参考基因组等。
最后,需要计算每个基因或转录本在各样本中的表达量,可以通过HTSeq、Cufflinks、RSEM等软件或者简单的读数对齐统计表达量。
2. 差异表达基因筛选:筛选出在不同样本间表达量差异显著的基因。
差异表达基因筛选是转录组分析的重要步骤之一,它可以将表达水平差异显著的基因筛选出来,对于研究基因的生物学功能、分子机制以及生物学意义都非常重要。
目前,常用的差异分析方法包括DESeq2、EdgeR、Limma、Cuffdiff等。
通过统计学方法和多重检验校正,可以筛选出不同样本中具有显著表达水平差异的基因。

基因组和转录组高通量测序数据分析流程和分析平台基因组和转录组高通量测序数据分析是生物信息学领域中的一个重要研究方向。
随着高通量测序技术的发展,获取大规模基因组和转录组数据已经成为可能。
通过对这些数据的分析,可以深入了解生物体内基因的表达和功能等相关信息。
本文将介绍基因组和转录组高通量测序数据分析的基本流程和常用的分析平台。
数据预处理是分析流程的第一步,主要包括测序数据的质控和去除低质量序列。
常用的质控工具包括FastQC和Trim Galore等,它们可以评估测序数据的质量和检测可能的污染。
在质控的基础上,可以使用Trimmomatic等工具去除低质量序列和适配体序列,保证后续分析的准确性和可靠性。
基因定量是分析流程的第二步,用于评估基因的表达水平。
常用的基因定量工具包括kallisto、Salmon和STAR等。
这些工具可以根据测序数据和已知的转录组序列,计算基因的表达水平。
基因定量的结果一般以表达矩阵的形式输出,该矩阵包含了每个样本中每个基因的表达值。
差异表达基因分析是分析流程的第三步,用于寻找基因表达水平在不同样本中存在显著差异的基因。
常用的差异表达基因分析工具包括DESeq2、edgeR和limma等。
这些工具可以对表达矩阵进行统计学分析,找出在不同样本之间具有显著差异的基因。
差异表达基因分析的结果一般以差异表达基因列表的形式输出。
富集分析是分析流程的第四步,用于寻找差异表达基因中富集的生物学功能或通路。
常用的富集分析工具包括GOseq、KEGG和enrichR等。
这些工具可以根据差异表达基因列表,基于GO注释和KEGG通路等数据库,计算差异表达基因在特定功能或通路上的富集度。
生物学注释是分析流程的最后一步,用于解释基因的功能和相关信息。
常用的生物学注释工具包括DAVID、GSEA和STRING等。
这些工具可以根据差异表达基因列表,提供关于基因功能、互作关系和代谢通路等信息。
除了上述基本流程外,还有一些附加的分析步骤,如差异剪接分析、外显子计数等。


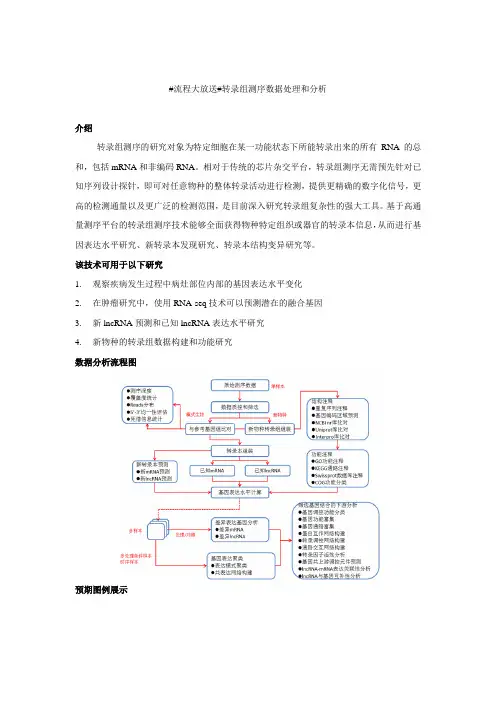
#流程大放送#转录组测序数据处理和分析
介绍
转录组测序的研究对象为特定细胞在某一功能状态下所能转录出来的所有RNA的总和,包括mRNA和非编码RNA。
相对于传统的芯片杂交平台,转录组测序无需预先针对已知序列设计探针,即可对任意物种的整体转录活动进行检测,提供更精确的数字化信号,更高的检测通量以及更广泛的检测范围,是目前深入研究转录组复杂性的强大工具。
基于高通量测序平台的转录组测序技术能够全面获得物种特定组织或器官的转录本信息,从而进行基因表达水平研究、新转录本发现研究、转录本结构变异研究等。
该技术可用于以下研究
1.观察疾病发生过程中病灶部位内部的基因表达水平变化
2.在肿瘤研究中,使用RNA-seq技术可以预测潜在的融合基因
3.新lncRNA预测和已知lncRNA表达水平研究
4.新物种的转录组数据构建和功能研究
数据分析流程图
预期图例展示
示例图1 差异表达基因筛选示例2 基因聚类分析heatmap图
示例3 差异基因互作网络图示例4 lncRNA、基因与上游共有miRNA网络图。
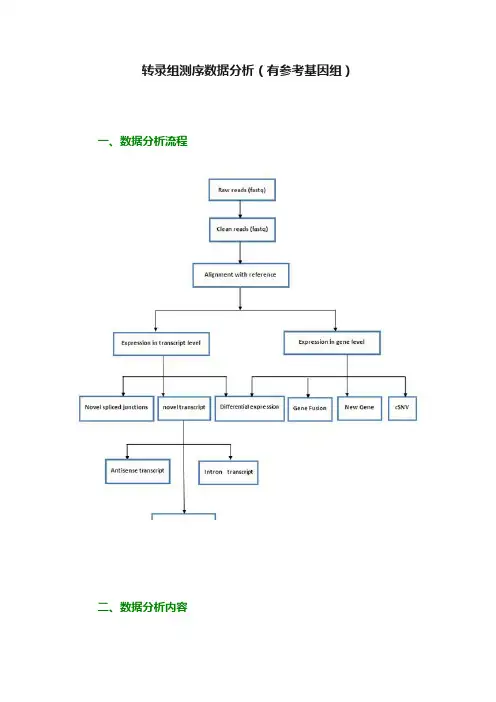
转录组测序数据分析(有参考基因组)一、数据分析流程二、数据分析内容1. 数据预处理目的:对原始测序数据进行一定程度的过滤。
原理:根据测序接头以及测序质量对原始的测序数据进行预处理,其中,测序质量Q与测序错误E之间的关系如下:结果:对预处理后质量以及碱基分布统计进行统计2. 比对基因组目的:将经过预处理的测序数据与参考基因组进行相似性比对。
原理:Burrower-Wheeler转换算法与splicing比对算法。
1)Burrower-Wheeler转换算法:由于测序数据量非常大,与整条基因组比对所需资源与时间是较为巨大的。
目前,我们采用Burrower-Wheeler(BWT)算法对基因进行建立索引、碱基压缩等过程,这样可以很大程度上加快比对速度,减少比对过程中所需资源。
2)splicing比对算法:即分段比对算法,当某条测序序列位于转录本剪切位点时,也就是这条序列同时属于两个外显子,如果将它与参考基因组进行比对,由于基因组两个外显子之间含有intron区,那么它将无法找到它合适的位置;但是应用分段比对算法就可以将这条测序序列分割变成多段子序列,然后应用这些段子序列与基因组进行比对,这样就可以找到它们真正的位置。
Vps28基因的一个分段比对的结果,蓝线连接的两端即为被分割的子序列,可见此种算法非常的适用于转录组测序。
结果展示:应用比对结果进行一些相关mapping统计,测序饱和度及测序5’,3’ bias统计。
Multi mapping,Unique mapping及Unique gene-body mapping统计。
饱和度分析,当reads达到一定测序量后,基因覆盖率基本达到饱和。
测序3’,5’偏好性统计,测序主要集中于基因bady区,两端偏向性较轻。
3. 基因表达水平研究目的:应用基因组比对结果进行基因定量。
原理:从指定物种基因模型(基因结构)中得到gene、exon、intron以及UTR等位置信息,通过基因组比对结果计算出在不用区域富集片段数目,然后应用RPKM/FPKM标准化公式对富集片段的数量进行归一化。




#流程大放送#转录组测序数据处理和分析
介绍
转录组测序的研究对象为特定细胞在某一功能状态下所能转录出来的所有RNA的总和,包括mRNA和非编码RNA。
相对于传统的芯片杂交平台,转录组测序无需预先针对已知序列设计探针,即可对任意物种的整体转录活动进行检测,提供更精确的数字化信号,更高的检测通量以及更广泛的检测范围,是目前深入研究转录组复杂性的强大工具。
基于高通量测序平台的转录组测序技术能够全面获得物种特定组织或器官的转录本信息,从而进行基因表达水平研究、新转录本发现研究、转录本结构变异研究等。
该技术可用于以下研究
1.观察疾病发生过程中病灶部位内部的基因表达水平变化
2.在肿瘤研究中,使用RNA-seq技术可以预测潜在的融合基因
3.新lncRNA预测和已知lncRNA表达水平研究
4.新物种的转录组数据构建和功能研究
数据分析流程图
预期图例展示
示例图1 差异表达基因筛选
示例2 基因聚类分析heatmap图
示例3 差异基因互作网络图
示例4 lncRNA、基因与上游共有miRNA网络图。
转录组结果解读转录调控研究部北京诺禾致源科技股份有限公司OUTLINE简介实验部分生物信息分析概述1转录组是指特定组织或细胞在某个时间或某个状态下转录出来的所有RNA的总和,主要包括mRNA和非编码RNA。
转录组研究是研究基因功能和结构的基础,对生物体的发育和疾病的发生具有重要作用。
RNA-seq技术流程主要包含两个部分,建库测序和数据分析。
2实验部分(RNA检测、建库、测序))•琼脂糖凝胶电泳:分析样品RNA完整性及是否存在杂质污染。
•NanoPhotometerspectrophotometer:检测RNA纯度(OD260/280及OD260/230比值)。
•Agilent 2100 bioanalyzer:精确检测RNA完整性。
链特异性文库优势:相同数据量下可获取更多有效信息;能获得更精准的基因定量、定位与注释信息5➢1、一般动物样品会有三条带:28S 、18S 、5S ,如果提取过程经过过柱处理或者利用CTAB+LiCl 方法提取,5S 可能较暗或者没有。
➢昆虫或者软体动物等样品只有1条比较明显的带,例如:牡蛎、果蝇、螨虫、蝗虫、蚊、蚕等➢2、植物样品有三条带:25S 、18S 、5S ,有些特殊物种或部位可能本身含条带比较多,如果条带清晰,也可初步判定合格➢3.原核生物中主要有5S 、16S 、23S rRNA叶片小鼠蚊动物植物原核RIN 5RIN 7RIN 8RIN 9RIN 4RIN 6RIN 10RIN 2RIN 1RIN 值范围示意图问与答文献要求OD260/OD230≥1.8,OD260/OD230如果小于2.0,则表明样品中被碳水化合物、盐类或有机溶剂污染;OD260/OD230合格的标准是多少呢?答:OD260/OD230≥2.0,且OD260/OD280≥2.0这说明RNA提取结果是相当好的,一般在1.8-2.1之间就说明RNA结果十分好,但是nanodrop的灵敏度没有2100好,因此我们主要根据2100检测结果来判定RNA是否合格,一般只要RIN值和RNA总量达到我们的判定标准的话,我们就会判为合格。
转录组高通量测序2010-11-22 09:48(第二代高通量测序技术-454)转录组即特定细胞在某一功能状态下所能转录出来的所有RNA的总和,是研究细胞表型和功能的一个重要手段。
与基因组不同的是,转录组的定义中包含了时间和空间的限定。
同一细胞在不同的生长时期及生长环境下,其基因表达情况是不完全相同的。
罗氏GS-FLX-Titanium第二代高通量测序仪平均读长超过400bp,在测序读长上遥遥领先于其它第二代高通量测序仪,使其成为转录组学研究的首选测序平台,已被广泛应用于基础研究、临床诊断和药物研发等领域。
一、罗氏454测序技术在环境微生物生态多样性研究中的突出优势体现在:(1)测序序列长,便于聚类拼接,可以对转录本进行从头组装(de novo assembly)。
(2)测序通量高,可以检测到低丰度转录本信息。
(3)可以对无基因组参考序列的新物种进行转录组测序,发现新的转录本和亚型。
(4)实验操作简单、结果稳定,可重复性强。
无需进行克隆的文库构建,双链cDNA连接454接头后可以直接进行测序,实验周期短。
(5)测序数据便于进行生物信息分析,可以进行基因差异表达分析、鉴定基因的可变剪切以及预测新基因。
二、美吉公司在环境微生物生态多样性研究中的突出优势体现在:(1)拥有自主实验室和高通量测序平台,可以根据客户要求灵活安排实验,实验周期短,取样方便,质量可靠。
(2)技术人员经验丰富,可以稳定地进行总RNA的提取和双链cDNA的合成,可以根据顾客要求第一时间提供实验方案。
(3)有专业的生物信息团队和大型计算机,可以为客户提供个性化的生物信息分析服务。
(4)开放式实验室,参与式服务。
客户不但可以参与整个实验过程,而且可以参与生物信息分析,提供最为增值的售后服务。
三、服务流程(1)客户提供样本背景信息、实验目的和实验预期。
(2)美吉公司设计实验方案,提供测序深度建议和生物信息分析建议。
(3)客户认可实验方案,双方签订项目合作协议。