转录组高通量测序转录组数据分析差异表因分析共66页
- 格式:ppt
- 大小:4.22 MB
- 文档页数:66

基因组和转录组数据的分析与解读随着基因组学和转录组学技术的发展,大量的数据被产生并储存起来,为研究基因功能、疾病诊断和治疗等方面提供了强有力的支持。
然而,这些数据的分析和解读需要大量的生物学知识和计算机科学技术,下文将介绍基因组和转录组数据的分析与解读的基本流程和方法。
基因组数据的分析与解读基因组的测序是指将一段DNA序列切割成数百万个小片段,并将这些小片段通过高通量测序技术测定其序列。
测序产生的序列数据需要进行整合、比对和注释。
基因组数据整合是指将数百万个小片段整合成原始DNA序列。
这个过程通常通过使用计算机程序来实现,比如Celera Assembler、SOAPdenovo和ABySS等。
这些程序根据DNA片段间的重叠信息来组装原始DNA序列。
基因组数据比对是指将测序产生的DNA序列与已知序列进行比对,从而确定它们在基因组上的位置。
这个过程通常使用BLAST、BWA和Bowtie等计算机程序来实现。
比对结果可以为基因的后续注释提供基础,同时也可以帮助进行基因组的各种功能分析。
基因组数据注释是指对基因组上的基因、转录本、启动子和调控元件等区域进行注释,从而确定它们的功能和作用。
这个过程可以通过使用基因组数据库、基因组注释软件和线上工具等来完成。
一般情况下,注释可以分为结构注释和功能注释两个部分。
结构注释包括基因边界的确定、外显子和内含子的识别和剪切位点的标注等;而功能注释则是对各种序列元件的功能进行预测和注释,比如基因调控区,非编码RNA序列,编码蛋白质序列等等。
转录组数据的分析与解读转录组学是对各种RNA分子的表达和调控机制进行研究的科学领域。
转录组数据分析可以帮助我们了解基因表达调控、寻找新型RNA分子和致病机制等。
转录组数据的分析与解读通常分为三个阶段:预处理、差异表达分析和富集分析。
转录组数据预处理包括了数据清洗、质量控制和对齐,以确保分析的数据质量。
数据清洗是针对测序数据的低质量和降解而设计的,目的是去除噪音和误差。
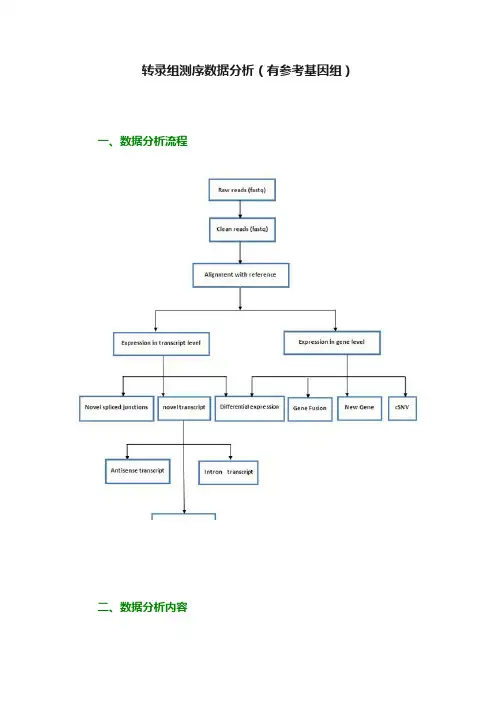
转录组测序数据分析(有参考基因组)一、数据分析流程二、数据分析内容1. 数据预处理目的:对原始测序数据进行一定程度的过滤。
原理:根据测序接头以及测序质量对原始的测序数据进行预处理,其中,测序质量Q与测序错误E之间的关系如下:结果:对预处理后质量以及碱基分布统计进行统计2. 比对基因组目的:将经过预处理的测序数据与参考基因组进行相似性比对。
原理:Burrower-Wheeler转换算法与splicing比对算法。
1)Burrower-Wheeler转换算法:由于测序数据量非常大,与整条基因组比对所需资源与时间是较为巨大的。
目前,我们采用Burrower-Wheeler(BWT)算法对基因进行建立索引、碱基压缩等过程,这样可以很大程度上加快比对速度,减少比对过程中所需资源。
2)splicing比对算法:即分段比对算法,当某条测序序列位于转录本剪切位点时,也就是这条序列同时属于两个外显子,如果将它与参考基因组进行比对,由于基因组两个外显子之间含有intron区,那么它将无法找到它合适的位置;但是应用分段比对算法就可以将这条测序序列分割变成多段子序列,然后应用这些段子序列与基因组进行比对,这样就可以找到它们真正的位置。
Vps28基因的一个分段比对的结果,蓝线连接的两端即为被分割的子序列,可见此种算法非常的适用于转录组测序。
结果展示:应用比对结果进行一些相关mapping统计,测序饱和度及测序5’,3’ bias统计。
Multi mapping,Unique mapping及Unique gene-body mapping统计。
饱和度分析,当reads达到一定测序量后,基因覆盖率基本达到饱和。
测序3’,5’偏好性统计,测序主要集中于基因bady区,两端偏向性较轻。
3. 基因表达水平研究目的:应用基因组比对结果进行基因定量。
原理:从指定物种基因模型(基因结构)中得到gene、exon、intron以及UTR等位置信息,通过基因组比对结果计算出在不用区域富集片段数目,然后应用RPKM/FPKM标准化公式对富集片段的数量进行归一化。

高通量测序数据分析总结引言高通量测序(high-throughput sequencing)是一种快速和高效地获取大量DNA或RNA序列信息的技术,被广泛应用于基因组学、转录组学和表观基因组学等领域。
随着高通量测序技术的发展,分析测序数据的能力也变得越来越重要。
本文将总结高通量测序数据分析的主要步骤和常用工具。
数据预处理在进行高通量测序数据分析之前,首先需要对原始测序数据进行预处理。
数据预处理的主要步骤包括:1.质量控制:使用质量控制工具(如FastQC)检查测序数据的质量,并去除低质量的读取。
2.去除接头序列:高通量测序数据通常会包含测序接头序列,需要使用工具(如Trimmomatic)去除这些序列。
3.低复杂度序列过滤:根据实验需求,可以使用工具(如Prinseq)过滤掉低复杂度的序列,以减少数据分析的噪音。
4.对reads进行比对:使用工具(如Bowtie、BWA)将reads与参考基因组或转录组进行比对,以获取比对到基因组或转录组的reads。
数据分析完成了数据预处理后,可以进行高通量测序数据的分析。
常见的数据分析任务包括:1.变异分析:通过比对到基因组的reads进行变异分析,识别单核苷酸变异(SNV)和小片段插入/删除(Indel)。
常用的工具有GATK、SAMtools 等。
2.转录本定量:利用比对到转录组的reads进行转录本定量分析,计算基因的表达水平。
常用的工具有Cufflinks、Salmon等。
3.差异表达分析:通过对比不同条件下的转录本表达水平,识别差异表达基因。
常用的工具有DESeq2、edgeR等。
4.GO/KEGG富集分析:通过对差异表达基因进行功能富集分析,探索这些基因的生物学功能和通路调控。
常用的工具有DAVID、Enrichr等。
5.其他分析:高通量测序数据还可以进行基因组装、转录因子结合位点分析、表观基因组学分析等。
结果展示高通量测序数据分析的结果可以通过各种方式展示,常用的包括绘制柱状图、散点图、热图、曲线图等。

高通量单细胞转录组测序数据分析流程下载提示:该文档是本店铺精心编制而成的,希望大家下载后,能够帮助大家解决实际问题。
文档下载后可定制修改,请根据实际需要进行调整和使用,谢谢!本店铺为大家提供各种类型的实用资料,如教育随笔、日记赏析、句子摘抄、古诗大全、经典美文、话题作文、工作总结、词语解析、文案摘录、其他资料等等,想了解不同资料格式和写法,敬请关注!Download tips: This document is carefully compiled by this editor. I hope that after you download it, it can help you solve practical problems. The document can be customized and modified after downloading, please adjust and use it according to actual needs, thank you! In addition, this shop provides you with various types of practical materials, such as educational essays, diary appreciation, sentence excerpts, ancient poems, classic articles, topic composition, work summary, word parsing, copy excerpts, other materials and so on, want to know different data formats and writing methods, please pay attention!高通量单细胞转录组测序数据分析流程在当前生物信息学领域中,高通量单细胞转录组测序数据的分析已成为了研究细胞功能、表型和组织结构的重要手段。

生物大数据技术在转录组差异表达分析中的使用技巧转录组差异表达分析是研究基因在不同条件下表达水平的变化,进而揭示基因功能及其与生物过程之间的关系的重要方法。
随着高通量测序技术的发展,生物大数据技术在转录组差异表达分析中的应用越来越广泛。
本文将介绍一些在转录组差异表达分析中使用生物大数据技术的技巧和注意事项。
首先,生物大数据技术在数据预处理方面发挥了重要作用。
转录组测序数据通常是以FASTQ文件的形式存在,然而,这些原始数据需要经过一系列的质控和预处理步骤,包括质量评估、质量控制、去除低质量序列、去除接头和连接序列等。
生物大数据技术提供了各种开源软件和工具,如FastQC、Trimmomatic和Cutadapt 等,可以高效地处理转录组测序数据,确保后续分析的可靠性和准确性。
其次,在差异表达分析中,生物大数据技术也提供了多种统计学方法和算法来评估基因表达的差异。
在差异表达分析中最常用的方法包括DESeq2、edgeR和limma等。
这些方法能够根据样本中的基因表达量,进行假设检验和统计模型拟合,找出在不同条件下表达差异显著的基因。
此外,还可以利用生物大数据平台提供的其他算法来进行聚类分析、主成分分析和绘制差异表达热图等,以进一步探索基因表达的模式和关系。
另外,生物大数据技术还能够提供基因功能注释和富集分析的相关信息。
转录组差异表达分析仅仅得到差异表达的基因列表是不够的,还需要了解这些基因的功能和相关的生物学过程。
生物大数据平台通常包含了丰富的基因功能注释信息,如基因本体、KEGG通路、GO功能和PPI等。
通过生物大数据技术的基因功能注释和富集分析工具,可以对差异表达基因进行功能分类、通路富集和相互作用网络分析等,从而深入理解差异表达基因的生物学意义。
此外,生物大数据技术还可以通过整合多组学数据来解析转录组差异表达。
转录组数据仅仅反映了基因的转录水平,而生物系统的功能往往需要多个层面的调控和互动。
生物大数据技术可以整合转录组数据与基因组、蛋白质组、代谢组等多组学数据,从而构建更全面的生物系统网络,揭示基因之间的调控网络和生物过程的整体调控机制。

转录组测序数据分析(无参考基因组)一、数据分析流程二、数据分析内容1. 数据预处理目的:对原始测序数据进行一定程度的过滤。
原理:根据测序接头以及测序质量对原始的测序数据进行预处理,其中,测序质量Q与测序错误E之间的关系如下:结果:对预处理后质量以及碱基分布统计进行统计2. UniGene拼接目的:将预处理后reads进行拼接,得到拼接结果。
原理:应用 de Bruijn graph path 算法对reads进行denovo拼接;对上一步的拼接结果,再用Hamilton Path算法拼接。
结果:UniGene序列,UniGene统计信息,序列长度分布图3. 数据库注释目的:对拼接得到的UniGene进行功能注释原理:通过blast+算法将拼接得到的UniGene序列与数据库进行比对结果:比对结果表格,物种分布统计和Evalue分布统计4. UniGene表达分析目的:UniGene定量分析。
原理:以UniGene为reference,分别将每个样本的reads进行reference mapping ,从而得到每个样本在每个UniGenes中的一个reads覆盖度,然后应用RPKM/FPKM标准化公式对富集片段的数量进行归一化。
RPKM:Reads Per Kilobase of exon model per Million mapped reads,公式下:FPKM:Fragments Per Kilobase of exon model per Million mapped reads,公式下:UniGene表达分布图,1X,5X分别为FPKM=1,FPKM=5分界点,可以大体观察到低表达,中表达以及高表达的比例关系UniGene样本间表达相关性散点图样本间表达差异程度的MA图,可以体现差异表达总体偏差5. UniGene表达差异分析目的:对定量结果进行统计检验分析,找出差异表达UniGene原理:双层过滤筛选差异基因FC值筛选:采用Fold-change(FC),表达差异倍数进行第一层此的差异基因筛选FDR检验:一般采用卡方检验中的fisher精确检验进行p值检验,采用Benjamini FDR(False discovery ratio)校验方法对p值进行假阳性检验,即,通过FDR显著性参数进行第二层次的差异基因筛选。

人参根、茎、叶转录组测序及差异表达基因分析目的:以五年生人参根、茎、叶为研究对象,利用高通量测序技术构建人参转录组数据库并筛选人参根与茎、叶差异表达基因,为进一步发现人参功能基因,阐明人参药效物质,选育优良品种等提供理论基础。
方法:运用改良的Trizol 法分别提取人参根、茎、叶总RNA,并采用琼脂糖凝胶非变性电泳及Agilent2100Bioanalyzer对其进行检测。
利用Illumina HiSeq2000系统进行转录组测序,使用Trinity软件做转录组从头组装,组装得到的序列使用Tgicl去冗余并进一步拼接,通过同源转录本聚类,得到最终的Unigenes。
不同样品得到的序列用聚类软件继续做拼接、去繁冗、并同源转录本聚类,最终得到不能再延长的非冗余All-Unigenes。
将非冗余Unigenes与nr、Swiss-Prot、KEGG和COG数据库做blastx比对(E value<10-5),取比对结果最好的蛋白确定最终的序列方向,获得基因注释信息,功能类别以及代谢通路等。
同以上数据库均比对不上的Unigene用ESTScan软件确定序列的方向。
根据数据库中基因表达量(FPKM值)筛选根、茎、叶高表达基因,根据基因表达量比值倍数的关系筛选根与茎、叶差异表达基因及非差异表达基因。
采用q-PCR方法对转录组数据库进行验证。
结果:1、改良Trizol法提取人参根、茎、叶总RNA,经琼脂糖凝胶电泳检测28S、18S条带清晰,亮度比例接近2倍;AgilentTechnologies2100Bioanalyzer检测O.D260/280在1.8~2.2之间,O.D260/230大于1.8,RIN>6.5,RNA总量>20μg,RNA符合建库标准。
2、运用HiSeq2000测序平台,双末端测序技术对序列进行拼接、去冗余后,每个样品平均获得4千多万条高质量的短序列。

干货整理转录组测序和分析,你需要知道的转录组测序及分析技术可以解决新基因的深度发掘、低丰度转录本的发现、转录图谱绘制、可变剪接的调控、代谢途径确定、基因家族鉴定及进化分析等各方面的问题;成为了广大科研工作者备受青睐的高通量测序技术之一。
转录组研究的应用领域十分广泛,适合研究组织特异性的、不同生长发育的、逆境胁迫下的、侵染转基因的、性状突变等材料。
转录组是在某一特定发育时期或某一生理条件下,细胞或组织内所有转录产物的集合,包括mRNA、lncRNA、small RNA、circle RNA等。
因此做转录组测序理论上可以研究各种长度范围的RNA序列,目前的常规技术包括mRNA测序、lncRNA测序、smallRNA测序。
那么问题来了,研究转录组如何下手?1根据研究对象,选择相应的建库策略(1)mRNA:可以通过富集polyA的方式来调取mRNA,进行建库测序;(2)lncRNA或lncRNA+mRNA:可以通过去rRNA试剂盒去除rRNA后进行建库测序;(3)circle RNA:可以通过消化线性RNA,再去除rRNA后进行建库测序;(4)small RNA:采用sRNA的建库策略,对18-40nt范围的sRNA进行切胶富集后建库测序。
2根据研究目的,选择不同的测序策略(1)了解不同样品间基因或sRNA的表达差异:选择SE(single end)测序即可,测序量10M reads以上;(2)进行基因的可变剪切、挖掘新基因、对现有基因的注释进行优化、检测基因融合等结构方面的分析:选择PE(pair end)测序,测序量则根据物种基因集合的大小来决定。
3基于转录组测序的主流研究手段(1)RNA-seq denovo:基于序列组装,用于从头构建某物种的转录本序列;(2)RNA-seq resequencing:对于已有参考基因的物种,进行基因定量、基因可变剪切、基因融合、新基因检测等分析;(3)lncRNA-sequencing:主要研究lncRNA的表达量,预测新的lncRNA及其功能;(4)sRNA sequencing:主要研究和分析small RNA序列,特别是miRNA的表达情况,并预测novel miRNA,miRNA靶基因分析等。

基于转录组测序的细胞与组织发育差异分析细胞和组织的发育差异一直是生物学研究的焦点,为了深入了解这些差异,科学家们采用了各种不同的策略。
其中,基于转录组测序的方法是近年来被广泛运用的一种技术,它可以分析细胞和组织中基因的表达情况,从而揭示出它们之间的差异。
本文将介绍转录组测序的基本原理和应用,以及当前在细胞和组织发育研究中的一些探索。
一、转录组测序的基本原理与流程转录组测序是一种高通量的技术,它能够同时测量所有基因表达的情况。
其基本原理是通过建立拷贝DNA文库和RNA-Seq测序技术来研究基因的表达情况。
转录组测序主要分为五个步骤:RNA提取、cDNA合成、文库构建、高通量测序和基因表达分析。
下面将分别介绍这五个步骤的具体内容。
(1)RNA提取RNA提取是转录组测序的第一步,目的是从不同的细胞或组织中分离出总RNA。
目前比较常用的方法是利用酚/氯仿法或RNA样品快速分离试剂盒来提取RNA。
其中酚/氯仿法是一种传统的RNA提取方法,它通过离心来分离出RNA。
但是其样品处理时间较长,且RNA的质量和数量难以保证。
而RNA样品快速分离试剂盒则是一种速度较快且操作简便的RNA提取方法,适用于各种样品、不同种类的组织和细胞。
(2)cDNA合成在RNA提取后,需要将其转录为cDNA才能进行测序。
cDNA合成有两种主要方法,即RT-PCR和切割酶法。
RT-PCR的优点是反应快,产量高,但存在一定的选择性和偏向性;切割酶法则不需要PCR,避免了PCR引起的偏差,但其产量相对较低。
(3)文库构建得到cDNA后,需要进行文库构建。
文库构建的目的是将样品中的cDNA片段插入到载体DNA中,并将其扩增。
扩增后的文库可以通过high-throughput测序技术进行高通量测序。
文库构建过程一般包括以下步骤:消化某些限制性内切酶来断裂cDNA片段,连接测序接头,进行PCR扩增,并通过凝胶回收方法获得合适大小的目标文库DNA。

转录组入门(7):差异表达分析PublicLibraryofBioinformatics理论基础:线性模型,设计矩阵和比较矩阵这部分内容最先在RNA-Seq Data Analysis的8.5.3节看到,刚开始一点都不理解,但是学完生物统计之后,我认为这是理解所有差异基因表达分析R包的关键。
基本上,统计课都会介绍如何使用t检验用来比较两个样本之间的差异,然后在样本比较多的时候使用方差分析确定样本间是否有差异。
当然前是样本来自于正态分布的群体,或者随机独立大量抽样。
对于基因芯片的差异表达分析而言,由于普遍认为其数据是服从正态分布,因此差异表达分析无非就是用t检验和或者方差分析应用到每一个基因上。
高通量一次性找的基因多,于是就需要对多重试验进行矫正,控制假阳性。
目前在基因芯片的分析用的最多的就是limma。
但是,高通量测序(HTS)的read count普遍认为是服从泊松分布(当然有其他不同意见),不可能直接用正态分布的t检验和方差分析。
当然我们可以简单粗暴的使用对于的非参数检验的方法,但是统计力不够,结果的p值矫正之估计一个差异基因都找不到。
老板花了一大笔钱,结果却说没有差异基因,是个负结果,于是好几千经费打了水漂,他肯定是不乐意的。
因此,还是得要用参数检验的方法,于是就要说到方差分析和线性模型之间的关系了。
线性回归和方差分析是同一时期发展出的两套方法。
在我本科阶段的田间统计学课程中就介绍用方差分析(ANOVA)分析不同肥料处理后的产量差异,实验设计如下肥料重复1 重复2 重复3 重复4A1 ... ... ... ...A2 ... ... ... ...A3 ... ... ... ... ...这是最简单的单因素方差分析,每一个结果都可以看成 yij = ai + u + eij,其中u是总体均值,ai是每一个处理的差异,eij是随机误差。
注:方差分析(Analysis of Variance, ANAOVA)名字听起来好像是检验方差,但其实是为了判断样本之间的差异是否真实存在,为此需要证明不同处理内的方差显著性大于不同处理间的方差。

转录组数据解读
嘿呀,朋友们!你们知道转录组数据不?这可真是个超级有趣的东西啊!就好比你有一堆拼图,而转录组数据就是这些拼图的碎片,等着你来把它们拼凑完整,搞清楚其中的奥秘。
比如说吧,我们可以把细胞想象成一个热闹的大舞台,基因就是舞台上的演员。
而转录组数据呢,就是这些演员在表演时的台词记录!是不是很形象?
你看啊,通过解读转录组数据,我们能知道在特定的情况下,哪些基因在“大声说话”,哪些在“悄悄嘀咕”。
这多有意思啊!就像我们在听一场精彩的戏剧,能了解每个角色的重要性和作用。
想象一下,要是我们对癌细胞的转录组数据进行解读,那不就相当于找到了攻克癌症的一把钥匙?这可太让人兴奋了吧!或者是研究植物的生长发育,了解它们在不同环境下的变化,那对农业的发展该有多大的帮助啊!
我记得有一次,和一群科学家朋友聊天,他们就特别激动地讨论着转录组数据解读的新发现。
一个说:“哎呀,这次发现的这个基因的表达变化,
简直像打开了一扇新的大门!”另一个接着说:“可不是嘛,这为我们进一步研究提供了重要线索呢!”然后大家都兴奋地七嘴八舌讨论起来。
转录组数据解读真的就像一个无尽的宝藏,等待着我们去挖掘。
它能让我们更深入地了解生命的奥秘,从微观的基因层面到宏观的生物现象。
它不仅是科学研究的重要工具,更是我们探索未知世界的强大武器啊!
所以啊,朋友们,别小看了这转录组数据,它可有着大魔力呢!让我们一起投入到这个神奇的领域中,去发现更多的精彩吧!我的观点就是:转录组数据解读有着巨大的价值和潜力,值得我们深入研究和探索。
转录组测序报告原始数据预处理转录组测序是一种用于研究生物体特定时间点或条件下的基因表达的方法。
在进行转录组测序之前,需要对原始数据进行预处理,以确保数据的准确性和可靠性。
数据质量控制在转录组测序之前,应对原始数据进行质量控制。
常用的质量控制工具包括FastQC和Trimmomatic等。
通过这些工具,可以检查数据的质量,如测序错误率、测序深度、GC含量等,并根据需求进行相应处理,如剔除低质量的reads,修剪低质量的碱基等。
数据预处理在进行转录组测序分析之前,需要对原始数据进行一系列的预处理步骤。
常见的预处理步骤包括:1.去除适配体序列:适配体序列是在测序过程中引入的一段DNA序列,如果不去除适配体序列,会影响后续分析的准确性。
2.去除低质量的reads:低质量的reads指的是测序质量较差的序列,可能由于仪器误差或实验操作等原因造成。
通过设置阈值,可以将低质量的reads剔除,以提高数据的质量。
3.去除rRNA序列:在转录组测序中,rRNA序列往往占据了大部分的reads,如果不去除rRNA序列,会影响后续对其他RNA类别的分析。
可以使用软件工具,如Bowtie和SortMeRNA等,去除rRNA序列。
4.剔除PCR重复序列:PCR重复序列是由于PCR扩增过程导致的序列复制,会引入偏差。
通过标记和去除PCR重复序列,可以减少这种偏差。
5.序列长度过滤:根据实验需求和研究题目的特点,设置合适的序列长度过滤阈值,过滤掉过短或过长的reads。
6.序列比对:将清洗后的reads与参考基因组或转录组进行比对,以获取每个reads在基因组或转录组中的定位信息。
表达量分析转录组测序可以用来研究细胞中的基因表达水平。
表达量分析是转录组测序中的重要环节之一,可以通过表达量分析来确定不同基因的相对表达水平以及在不同条件下的差异表达。
转录本定量在表达量分析中,首先需要进行转录本定量。
转录本定量是指对转录组测序得到的reads进行比对,并根据比对结果计算每个转录本在给定样本中的表达量。
#流程大放送#转录组测序数据处理和分析
介绍
转录组测序的研究对象为特定细胞在某一功能状态下所能转录出来的所有RNA的总和,包括mRNA和非编码RNA。
相对于传统的芯片杂交平台,转录组测序无需预先针对已知序列设计探针,即可对任意物种的整体转录活动进行检测,提供更精确的数字化信号,更高的检测通量以及更广泛的检测范围,是目前深入研究转录组复杂性的强大工具。
基于高通量测序平台的转录组测序技术能够全面获得物种特定组织或器官的转录本信息,从而进行基因表达水平研究、新转录本发现研究、转录本结构变异研究等。
该技术可用于以下研究
1.观察疾病发生过程中病灶部位内部的基因表达水平变化
2.在肿瘤研究中,使用RNA-seq技术可以预测潜在的融合基因
3.新lncRNA预测和已知lncRNA表达水平研究
4.新物种的转录组数据构建和功能研究
数据分析流程图
预期图例展示
示例图1 差异表达基因筛选
示例2 基因聚类分析heatmap图
示例3 差异基因互作网络图
示例4 lncRNA、基因与上游共有miRNA网络图。
梁山慈竹高通量转录组测序及差异表达基因分析王身昌;胡尚连;曹颖;徐刚【摘要】分析梁山慈竹及其体细胞突变体的转录组,挖掘功能基因并对其差异表达基因进行筛选和分析,为梁山慈竹遗传改良提供理论依据。
利用 RNA-Seq 技术进行转录组测序,对测序结果进行 de novo 拼接和功能注释;并对差异表达基因进行筛选及 COG、GO、KEGG 数据库中进行比对注释,此外,基于 Swiss-Prot 功能注释结果,分析纤维素和木质素相关功能基因的表达量差异。
测序结果表明,共获得86575631条 reads,de novo 组装得到84741条 unigenes,共有49829条被 Nr、COG、GO、KEGG、Swiss-Prot 注释。
从梁山慈竹实生植株(对照)和体细胞突变体 No.30这2个测序样本中,筛选出3572条差异表达unigenes,757条差异表达 unigenes 在 COG 分类体系中具有详细的蛋白功能释义,2213条差异表达 unigenes 在 GO 数据库具有功能定义,385条unigenes 被注释到94条 KEGG Pathways 中。
纤维素合成相关纤维素合酶、过氧化物酶、泛素连接酶和热休克蛋白在梁山慈竹体细胞突变体 No.30中表达量升高,木质素合成相关 MYB4、4-香豆酸 CoA 连接酶、肉桂醇脱氢酶、肉桂酰-CoA 还原酶和漆酶在突变体中表达量降低。
提供了全面的梁山慈竹转录组信息,获得了一批在梁山慈竹纤维素和木质素生物合成过程中有重要功能的基因序列。
%The transcriptome from the shoots of Dendrocalamus farinosus and its somatic mutant was sequenced using the RNA-Seq technology to elucidate its functional gene and analyze its differential expressed gene,providing a theoretical basis for its genetic improvement.The sequencing data was assembled by de novo assembly and the dif-ferential expressed gene screened was annotated in COG,GO and KEGG database.Inaddition,the differential ex-pression of gene related to cellulose and lignin biosynthesis was analyzed,basing on the Swiss-Prot function annota-tion.The sequencing results showed that a total of 86 575 631 reads were produced and assembled into a total of 84 741 unigenes by denovo,among which 49 829 were annotated in Nonredundantprotein,Cluster of Orthologous Groups of proteins,Gene Ontology,Kyoto Encyclopedia of Genes and Genomes and Swiss-Prot database.Besides,a total of 3 572 differential expressed unigenes were identified from the plant of seeds (CK)and the somatic mutant No.30.Of these differential expressed genes,757 unigenes had a detailed protein functions in the COG classification system,2 213 unigenes had the function definition in the GO database,385 unigenes were annotated to 94 KEGG Pathways.The expression of genes encoding CesA,Prx,Ubiquitin-conjugating enzyme and Heat shock proteins in-creased in somatic mutant No.30,while,the expression of genes encoding MYB4,4CL,CAD,CCR and LAC de-creased.Our data provide the most comprehensive transcriptomic resource for Dendrocalamus farinosus and the tran-script sequences with important function related to cellulose and lignin biosynthesis were found,which provide the most precious information resources for the further research on bamboo.【期刊名称】《华北农学报》【年(卷),期】2016(031)003【总页数】7页(P65-71)【关键词】梁山慈竹;体细胞突变体;转录组;差异表达基因【作者】王身昌;胡尚连;曹颖;徐刚【作者单位】西南科技大学植物细胞工程实验室,四川绵阳 621010; 四川省生物质资源利用与改性工程技术研究中心,四川绵阳 621010;西南科技大学植物细胞工程实验室,四川绵阳621010; 四川省生物质资源利用与改性工程技术研究中心,四川绵阳 621010;西南科技大学植物细胞工程实验室,四川绵阳 621010; 四川省生物质资源利用与改性工程技术研究中心,四川绵阳 621010;西南科技大学植物细胞工程实验室,四川绵阳 621010; 四川省生物质资源利用与改性工程技术研究中心,四川绵阳 621010【正文语种】中文【中图分类】Q78转录组(Transcriptome)能够从整体水平研究基因功能以及基因结构,揭示特定生物学过程中的分子机理[1]。
第一章转录组及转录组测序第一节前言1953年,沃森与克里克对DNA双螺旋结构的精确描绘开创了生命科学的黄金时代,随后如火如荼的开展起来的人类基因组计划建立起庞大复杂的基因组数据库使人类对了解生命本源和控制生命进程燃起无限憧憬。
随着越来越多的基因测序工作渐渐完成,一本“写满生命密码的天书”呈现在我们面前, 然而,接下来的问题更纷扰而至:1) 这些基因有什么功能?2) 不同的基因参与了哪些细胞内不同的生命过程?3) 基因的表达是如何调控的呢?4) 基因与基因产物之间是如何相互作用的呢?5) 相同的基因在不同的细胞内的表达水平有差异吗?6) 相同的基因处于疾病和治疗状态下的表达水平会有哪些改变?如何读懂这本“天书”是目前横亘在科学家们面前严峻的挑战。
因此,在人类基因组项目后,转录组学,蛋白组学,代谢组学等组学不断涌现,生命科学研究已经跨入后基因组时代。
其中,转录组学作为一个率先发展起来的学科是研究细胞表型和功能的一个重要手段,转录组高通量测序技术开始在生物学前沿研究中得到了广泛的应用。
第二节转录组(transcriptome)与转录组学(transcriptomics)读懂基因组这本“天书”,最先要研究清楚基因是怎么表达的。
所谓基因表达,是指将基因携带的遗传信息转变为可辨别的表型的整个过程。
基因表达的第一步, 也即基因表达调控的关键环节,是以DNA为模板合成RNA的转录过程。
转录后的所有mRNA的总称即转录组。
由转录组延伸出来一门学科即转录组学,它是分子生物学的分支,负责研究在单个细胞或一个细胞群的特定细胞类型内所产生的mRNA分子,是从RNA层次研究基因表达的情况。
第三节转录组研究的重要性转录组是连接基因组遗传信息与生物功能的蛋白质组的纽带,转录水平的调控是最重要也是目前研究最广泛的生物体调控方式。
转录组的研究比基因组的研究能给出更高效的有用信息。
比如,人类基因组包含有30亿个碱基对,其中大约只有5万个基因转录成mRNA分子,而转录后的mRNA仅部分被翻译生成功能性的蛋白质。
高通量测序数据差异分析(DESeq2)今天我们来学习R语言DESeq2做差异分析,第一次我推送这个差异分析到现在已经过去一年多了,我重新排版,更加了一些感悟,重新推送给大家。
基于高通量测序数据的差异分析,为了矫正测序平台,批次,深度等差异,所以这里做DEsep2差异分析。
这个包适用于:高通量数据分析过程中,基于count数据,对其进行标准化处理,并对两个样本的差异做定量比较。
注意这里是基于read数进行分析的,卖的是标准化方法。
说道安装DEsep2这个包,这段时间在ubuntu中安装更加困难,还好最后都解决了。
这个包绝对算得上的较难安装的包之一。
当然现在我目前安装最为困难的包是microbiomeSeq,大家有时间可以试试,做扩增子的同学可以看看这个包,是有作者的心意在里面的。
#安装这个包,安装过程不太顺利,请多试几次,还是没问题的,我也没有出现意料之外的问题source("https:///biocLite.R")biocLite("DESeq2")library(DESeq2)读取我们的数据,这里使用两个文件,一个是分组文件,另一个是OTU表格。
有的人曾问过,这个OTU表格需要抽平吗?这里我直接回答:不需要。
# 读入实验设计,Qiime的mapping文件删去第一行的“#”即可使用design =read.table("map_lxdjhg.txt", header=T, s= 1,sep="\t")head(design)# 读取OTU表,全部otu表没有抽平,基于count的数据,不可用相对丰度数据otu_table =read.delim("otu_table.txt", s= 1,sep="\t",header=T,s=F)#做判别,做排序为了使变量可以一一对应上idx =rownames(design) %in% colnames(otu_table)design = design[idx,]#我们后面利用sub_design文件做分组文件sub_design =design[idx,]#我们后面利用count文件做差异分析文件count =otu_table[, rownames(sub_design)]#这里我们将count转化为矩阵,当然可以不用转化count=as.matrix(count)head(count)第一步构建两个矩阵,第一个OTU矩阵,第二个分组矩阵dds <-DESeqDataSetFromMatrix(countData = count,colData =sub_design,design = ~ SampleType)countData:即为差异分析otu表格,格式为行名otu名称,列名变量名为样品名colData:即为分组文件;design:即为分组文件中指定分组的列的列名;第二步dds2 <-DESe q(dds)#直接用DESeq函数即可resultsNames(dds2)#这里我们指定需要比较的两组样本# 将结果用results()函数来获取,赋值给res变量,这里我设定显著性阈值为alpha=0.05res <- results(dds2,contrast=c("SampleType","G0", "GC1"),alpha=0.05)#看一下结果的概要信息summary(res)提取结果进行保存#这里我们可以更据结果文件提取我们需要的行,使用subset函数,也是也个很有用的函数WT<-subset(res,padj< 0.05& (log2FoldChange > 1| log2FoldChange < -1))#当然可以只得到想要的OTU名称WT2 <s(WT)# res就是最后的显著性比较的文件,这里我们按照显著性排序res <-res[order(res$padj),]#将差异和OTU相对丰度做一个合并wt3<- merge(as.data.frame(res),as.data.frame(counts(dds2,normalize =TRUE)),by="s",sort=FALSE)head(wt3)# 得到csv格式的差异表达分析结果write.table(wt3,"otu差异统计表格GC1-G0.txt",quote= FALSE,s = T,s = T,sep = "\t")这里提到了相对丰度提取的命令## 获得normalize的countdds <-estimateSizeFactors(dds)wt6 <-counts(dds, normalized=T)head(wt6)这是上面我们合并采用的命令counts(dds2,normalize=TRUE)学习永无止境,分享永不停歇!。