基于文献挖掘和基因共表达网络的基因与疾病关联分析
- 格式:doc
- 大小:12.53 KB
- 文档页数:2

加权基因共表达网络分析结合机器学习算法筛选与腹主动脉瘤免疫相关的关键基因杨树;张娣;谢春杨【期刊名称】《血管与腔内血管外科杂志》【年(卷),期】2024(10)3【摘要】目的探讨腹主动脉瘤(AAA)免疫相关的关键基因。
方法从基因表达综合数据库(GEO)中获取AAA组织与健康人群正常腹主动脉组织的转录组数据,通过单样本基因集富集分析(ssGSEA)计算免疫细胞浸润分数,使用加权基因共表达网络分析(WGCNA)结合机器学习算法筛选与AAA免疫浸润相关的关键基因,使用STRING数据库进行蛋白-蛋白互作分析,采用受试者工作特征(ROC)曲线分析目的基因对于AAA的诊断能力。
结果与健康人群腹主动脉组织相比,AAA组织中活化的CD4^(+)T细胞浸润分数升高,差异有统计学意义(P﹤0.05)。
WGCNA分析获得1215个与活化的CD4^(+)T细胞浸润相关的基因,差异基因表达分析得出990个AAA组织和健康人群正常腹主动脉组织表达差异的基因,将差异基因表达分析得到的基因与WGCNA中所得的基因进行交集后得到282个与CD4^(+)T细胞浸润相关的差异基因。
将282个差异基因进行基因本体论(GO)、京都基因与基因组百科全书(KEGG)富集分析,生物过程(BP)富集分析表明,这些基因与有机化合物氧化的能量衍生、细胞呼吸、线粒体呼吸链复合物组装、烟酰胺腺嘌呤二核苷酸(NADH)脱氢酶复合物装配和线粒体呼吸链复合体I装配等生物过程有关;分子功能(MF)富集分析表明,这些基因与氧化还原相关的分子学功能有关;细胞成分(CC)富集分析表明这些分子表达与线粒体组分相关。
KEGG富集分析显示,这些基因与神经系统疾病、非酒精性脂肪肝、氧化磷酸化等信号通路有关。
通过向量机-相对误差过滤(SVM-REF),LASSO逻辑回归和随机森林模型3种机器学习算法从282个与活化的CD4^(+)T细胞浸润相关的差异基因中获得了4个Hub基因(VCAN、CUTL1、TRAPPC4和LOC646782)。

基因共表达网络的构建及其相关性分析近年来,随着高通量技术的发展,基因数据的产出速度也在不断加快。
然而,单个基因的研究往往无法发现复杂疾病背后的机制,而对基因共表达网络的构建及其相关性分析能够探索基因之间的相互作用,从而揭示得疾病的本质。
基因共表达网络是指通过计算基因表达量的相似性,将基因相互联系起来形成的网络。
与传统的研究方式不同,基因共表达网络将基因看做一个整体,旨在研究基因的相互影响,从而更好地理解生物系统的复杂性。
当前,基因共表达网络已被广泛地应用于多种研究领域,比如疾病筛选、药物开发、基因调控网络的重构等。
构建基因共表达网络的基本步骤包括数据预处理、基因表达数据标准化、基因表达相关系数计算、筛选相关性达到一定标准的基因,并将它们构成一个网络图等。
常用的数据预处理方法包括质量控制、归一化、去除批次效应等。
目前主要有Pearson相关系数、Spearman相关系数和互信息等方法用于基因表达的相关系数计算。
在筛选相关性较高的基因时,常用的方法有阈值法、P值法、False Discovery Rate(FDR)法或者公认的基因相关模型等。
基因共表达网络分析不仅关注单个基因,更重视整体上基因之间的协同作用与相互关联,需要从全局的角度去探究基因网络中的基因间相互作用关系。
基因网络分析的主要内容包括度数分布、节点中心性、聚类分析和模块检测。
节点度数分布是指节点在整个网络中的连接数分布状况,通常用来表征网络的复杂性和稳健性。
而节点中心性能够评估各个节点在网络中的重要性,并说明节点在整个网络结构中所处的位置。
常见的节点中心性指标包括度中心性、介数中心性、接近中心性等。
聚类分析是基于节点的相似性来讲整个网络划分成若干个子网络并对其进行进一步分析的一种方法。
聚类分析可以使得相似的基因或样本聚集在一起,方便对其进行进一步的生物学研究。
常见的聚类算法包括Hierarchical Clustering和K-Means 算法等。

大学生生物信息学考试模拟题及解析一、单选题(每题 3 分,共 30 分)1、生物信息学中,用于分析 DNA 序列的常见软件是()A BLASTB ClustalWC Primer PremierD MEGA2、以下哪种数据库主要存储蛋白质结构信息()A GenBankB PDBC UniProtD SWISSPROT3、在基因预测中,开放阅读框(ORF)是指()A 从起始密码子到终止密码子的一段序列B 具有特定功能的一段基因序列C 编码蛋白质的基因序列D 以上都不对4、进行系统发育分析时,常用的构建进化树的方法是()A 邻接法B 最大简约法C 最大似然法D 以上都是5、以下哪种算法常用于序列比对()A 动态规划算法B 贪心算法C 分治法D 回溯算法6、生物信息学中,用于分析基因表达数据的常用方法是()A 聚类分析B 回归分析C 方差分析D 以上都是7、以下哪个不是常见的生物信息学文件格式()A FASTAB GenBankC PDBD CSV8、在蛋白质序列分析中,用于预测蛋白质二级结构的方法是()A 同源建模B 从头预测C 基于机器学习的方法D 以上都是9、进行基因功能注释时,常用的数据库是()A GOB KEGGC ReactomeD 以上都是10、以下哪种技术可以用于大规模测序()A Sanger 测序B 二代测序C 三代测序D 以上都是答案及解析:1、答案:A解析:BLAST(Basic Local Alignment Search Tool)是用于比较生物序列相似性的工具,常用于分析 DNA 序列。
ClustalW 主要用于多序列比对;Primer Premier 常用于设计引物;MEGA 用于构建进化树。
2、答案:B解析:PDB(Protein Data Bank)是主要存储蛋白质结构信息的数据库。
GenBank 主要存储核酸序列;UniProt 和 SWISSPROT 主要存储蛋白质序列信息。

AI医疗制定个性化用药方案随着人工智能(AI)技术的迅猛发展,医疗领域也开始广泛应用AI技术,其中之一就是AI医疗在制定个性化用药方案方面的应用。
个体差异在用药效果上存在着显著影响,因此,提供个性化的用药方案对患者的治疗效果起着重要作用。
本文将探讨AI医疗制定个性化用药方案的意义、现状以及未来发展趋势。
一、AI医疗制定个性化用药方案的意义个性化用药方案是根据患者的特定病况、基因信息、生活习惯等综合因素制定的用药方案。
通过AI医疗制定个性化用药方案,可以最大限度地减少患者的用药风险,提高治疗效果。
其意义主要体现在以下几个方面:1. 提高用药效果:每个人的基因构成都存在差异,因此对于相同的疾病,同样的药物对不同的个体可能会产生不同效果。
通过分析患者基因信息,AI医疗可以制定针对个体的用药方案,提高用药效果。
2. 避免药物不良反应:患者在用药过程中可能会出现药物不良反应。
通过AI医疗可以预测出患者可能会出现的不良反应,从而避免不必要的风险。
3. 降低医疗成本:个性化用药方案的制定可以提高用药的针对性,从而减少不必要的试错过程,降低医疗成本。
二、AI医疗制定个性化用药方案的现状目前,AI医疗在制定个性化用药方案领域已经取得了一定的进展。
以下是目前AI医疗制定个性化用药方案的主要应用方式:1. 基于患者数据的分析:AI医疗通过分析患者的临床表现、病历记录、实验室检查结果等多种数据,建立患者的健康档案,进而制定个性化的用药方案。
2. 基于基因信息的分析:通过分析患者的基因信息,AI医疗可以预测患者对某种药物的代谢能力、药物吸收率等,进而制定个性化的用药方案。
3. 基于文献数据库的分析:AI医疗可以通过分析大量的文献数据库,挖掘出药物与基因之间的关联性,从而制定更加个性化的用药方案。
三、AI医疗制定个性化用药方案的未来发展趋势随着技术的不断进步和数据的不断积累,AI医疗在制定个性化用药方案方面有着广阔的发展空间。

基因共表达网络的构建与分析基因共表达是指一组基因在特定条件下的表达水平高度相关。
与其他无关的基因相比,共表达基因具有更高的生物学意义。
举例来说,同属于某个代谢途径或信号通路的基因,可能会表现出高度的共表达,这种关系可以用共表达网络的形式来呈现。
构建基因共表达网络的一般步骤包括生物样本采集、基因表达分析、基因表达矩阵转化和网络构建等。
其中,网络构建是整个过程中最关键和最具挑战性的步骤。
一般来说,网络构建需要解决以下几个问题:第一,如何选择构建网络的方法和算法? 第二, 基因的选择、表达和相关性分析等因素对网络的构建产生了哪些影响? 第三,如何有效的验证网络的可靠性和生物学意义?网络构建方法网络的构建方法主要有两种:基于概率论的方法和基于相关性的方法。
基于概率的方法包括了贝叶斯网络、高斯图模型、动态贝叶斯网络等;而基于相关性的方法常用的则是Pearson相关系数、Spearman秩相关系数、互信息等。
其中,基于相关性的方法相对于基于概率的方法,具有计算简单、易于理解和解释结果等优点。
不过,基于相关性的方法也存在着一些问题。
例如,相关性是基于样本的,存在着一些假阳性和假阴性;同时,相关性未必能够反映出基因相互作用的复杂性和多样性。
基因的选择和表达矩阵对于网络的构建,基因的选择和表达矩阵也是非常重要的环节。
选择所有基因来构建网络的策略是不现实的,因为它既会导致计算的复杂性,而且还可能掩盖了实际情况中的关键基因。
相反,应该根据预期的研究目标,选择一些关键基因进行构建。
选择的基因可以根据文献报道、代谢途径或功能、公共数据库的注释信息、基因元数据或从已有的表达数据集中挖掘等方式来确定。
对于表达数据的矩阵转换,一般将基因表达谱的数据矩阵进行标准化或归一化,以消除处理过程中的误差并提高数据的可靠性。
一些方法也可以来消除样品效应和探针效应等干扰。
网络分析和验证网络构建完成后,需要进行网络分析和验证。
网络分析是从网络中提取关键基因和关键信息的过程。

生物信息学中的基因共表达网络分析研究近年来,生物信息学的应用越来越广泛,其中基因共表达网络分析是研究基因之间相互作用和调控关系的重要手段。
本文将介绍基因共表达网络分析及其在生物学研究中的应用。
一、基因共表达网络分析的概念和方法基因共表达网络分析是通过分析基因表达数据来构建基因共表达网络,即将具有相似表达模式的基因连接起来形成一个网络。
在这个网络中,每个基因被看作是一个节点,基因之间的相似度则用各种计算方法来度量。
基因的表达模式可以是基于时间、组织或环境等条件的表达模式。
经过这样的计算,就可以将所有基因分成若干个模块(module),每个模块中的基因具有相似的表达模式。
在网络构建完成后,可以对模块内的基因进行聚类分析,得到具有类似功能或相似表达模式的基因集群。
这些基因集群代表了基因相互作用的一部分,可以揭示基因在共同参与生物学过程时的功能。
二、基因共表达网络分析在生物学研究中的应用基因共表达网络分析已经被广泛应用于生物学研究中,例如:1.鉴定功能相关基因通过构建基因共表达网络,可以鉴定与某个生物过程相关的基因模块,并对模块中的基因进行功能分析。
这种方法可以为生物学研究提供有力的参考。
2.预测新的基因功能基因共表达网络分析可以通过结合已知功能的基因和未知功能的基因来预测未知基因的功能。
3.挖掘基因互作网络基因共表达网络分析可以揭示基因之间的互作关系,进而构建基因互作网络。
这种方法可以为药物靶点预测和疾病诊断提供重要的信息。
三、基因共表达网络分析的一些应用案例1.构建植物中的基因共表达网络基因共表达网络分析已经在植物学研究领域得到了广泛应用。
例如,在一篇研究中,研究人员构建了植物中的基因共表达网络,通过对网络中的模块进行聚类分析,鉴定了具有调控光合作用和抗氧化系统等生物学功能的基因模块。
2.揭示异色素在人类癌症中的功能满足在一项研究中,研究人员利用基因共表达网络分析的方法,在人类癌症中揭示了异色素(Irisin)通过对胰岛素抗性和代谢疾病的调节而发挥重要作用的机制。



基因共表达——基因共表达网络分析Gene co-expression(基因共表达)是一种使用大量基因表达数据构建基因间的相关性,从而挖掘基因功能的一类分析方法。
在很多情况下,有着相似行为/变化的物质,会存在着一定的联系。
在生物中,存在于同一个通路的基因在表达值上,会表现出共表达的趋势(co-expression patterns),通过这个特性可以进行基因功能(一般的基因注释是通过序列的相似性,而通过基因共表达可以进行功能同源基因的注释)、module等的挖掘,这是一种guilt-by-association的思想。
module 指的是在共表达网络中,一组相互连接紧密的node(基因),一般认为一个module中的基因在功能上是相关的。
通常通过设定阕值来进行寻找在共表达分析中,有两种有效的策略Guide-genes 和Non-targeted:image.pngGuide-genes策略是,先找到感兴趣的基因/通路的共表达基因,进行可视化,然后再加入另外的基因,看两次加入的基因之间存在怎样的关系,以此来基因的关系。
Non-targeted 策略与Guide-genes不同,它没有感兴趣的基因,所以它从头构建了所有表达数据的共表达网络,然后寻找其中的module。
然后然后去探究module的功能。
Non-target策略中检测module的方法image.pngmodule的检测是基因共表达网络分析中非常重要的一步,一般有两种方法:top-down(A)和 bottom-up(B)top-down方法是将两个相互连接的区域进行分离,每个区域各自是具有比较高的Network density,区域之间的连接较区域内的连接是少比较多的。
这样便可以将两个区域分开,各自当成一个module。
bottom-up方法是在一个单独的区域中,不断加入单个基因,检测这个单基因是否与区域内大部分基因具有共表达关系,判断是否能融于区域中。

基因表达调控网络的解析及其应用基因表达调控网络是生物学研究中一个重要的研究领域,其在生物学、医学、生物工程等领域都有着广泛的应用。
基因表达调控网络包括基因、非编码RNA、蛋白质、代谢物等多个分子水平,涵盖了调控、信号传递等多个生命网络。
本文将着重介绍基因表达调控网络的解析及其应用。
一、基因表达调控网络解析1、基因表达调控网络的建立基因表达调控网络的建立可以通过多个方法,例如: 基于转录因子/靶基因数据、基于组蛋白修饰数据、基于DSB-seq、DNase-seq、ATAC-seq等数据,以及基于单细胞转录组学数据等。
2、调控元件预测调控元件预测可以通过多种方法,例如: 基于转录因子结合位点、组蛋白修饰、DNA甲基化等方法,以及通过机器学习和深度学习等方法。
3、基因共表达网络建立基因共表达网络可以通过表达物整合、数据聚类等多种方法来建立。
基因共表达网络的建立可以揭示不同的调控子网络、遗传路径。
4、基因调控网络的建立基因调控网络可以通过结合基因表达数据、转录因子结合位点、基因共表达网络等多个数据来建立。
基因调控网络的建立可以揭示不同的调控子网络、遗传路径。
二、基因表达调控网络的应用1、疾病分类基因表达调控网络的疾病分类包括多种类型,如: 基于单细胞转录组学数据的多种疾病分类,基于癌症的疾病分类等。
2、预测调控元件预测调控元件的方法可以通过基于转录因子结合位点等,通过机器学习和深度学习等方法来预测。
3、分析基因表达调控网络和疾病相关的谷物调控基因表达调控网络和疾病相关的谷物调控,包括基因组选择、谷物转录因子和非编码RNA的功能调查等,验证了基因组和调控过程的生物学意义,使我们更好的理解了其组织发育,并为其改良和研发金利草提供理论依据。
4、新药研发基因表达调控网络在新药研发中的应用,包括通过基因调控网络发现新药靶标、通过基因共表达网络发现新药靶标、通过计算化学和多肽库设计等方法设计药物等。
结论基因表达调控网络是一个复杂的生物网络,其的建立和应用是生物学研究中一个重要的研究领域,解析基因表达调控网络可以帮助我们更好的理解生物学过程,并为其在医学、生物工程等领域的应用提供了理论依据。
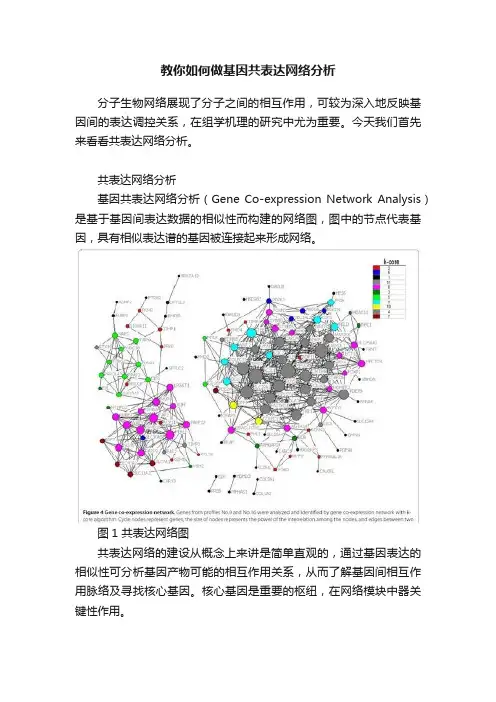
教你如何做基因共表达网络分析分子生物网络展现了分子之间的相互作用,可较为深入地反映基因间的表达调控关系,在组学机理的研究中尤为重要。
今天我们首先来看看共表达网络分析。
共表达网络分析基因共表达网络分析(Gene Co-expression Network Analysis)是基于基因间表达数据的相似性而构建的网络图,图中的节点代表基因,具有相似表达谱的基因被连接起来形成网络。
图1 共表达网络图共表达网络的建设从概念上来讲是简单直观的,通过基因表达的相似性可分析基因产物可能的相互作用关系,从而了解基因间相互作用脉络及寻找核心基因。
核心基因是重要的枢纽,在网络模块中器关键性作用。
案例分析下面以文献为例,来看看如何用共表达网络分析找出关键的节点基因。
文献信息Genes related to the very early stage of ConA-induced fulminant hepatitis: a gene-chip-based study in a mouse model 相关基因:ACSL1、MPDZ相关疾病:Liver Failure样本:GSE17184PMID: 20398290 IF: 3.867这篇文章主要研究爆发性肝炎在早期阶段的病理机制,作者在寻找病变过程中起主导作用的基因群及关键基因时,采用ConA 诱导的小鼠肝炎模型,选取了0h,1h,3h,6h四个时间点做表达谱分析。
用ANOVA筛选得到了1473个差异表达基因(p<0.05,FDR<0.05)。
为进一步筛选出显著变化的基因群,作者将差异基因进行趋势分析,从中得到了10个显著变化的趋势模型(p<0.05)并将最显著变化的NO.9和NO.16趋势模型中的基因进行共表达网络分析,来判断哪些基因在爆发性肝炎早期阶段起主导作用。
在以k-core值分的子网络中(k-core值越大表明子网络越处于核心的地位),作者筛选得到了k-core值最高为11的子网络中的26个核心基因。
生物大数据技术中的基因共表达网络分析方法介绍基因共表达网络(gene co-expression network)是生物大数据技术中一种重要的分析方法,它可以帮助研究人员理解基因之间的相互作用和调控机制。
本文将介绍基因共表达网络的分析方法及其在生物学研究中的应用。
首先,我们需要了解什么是基因共表达网络。
基因共表达网络是根据基因表达谱中基因的相关性构建起来的一个图形化模型。
在生物体内,基因的表达水平受到多种因素的调控,包括遗传、环境和生理等。
基因共表达网络通过分析不同样本中基因表达量的变化,并找出表达模式相似或相关的基因,将它们连接起来形成一个网络结构。
该网络可以帮助我们发现潜在的基因功能和生物过程。
基因共表达网络的构建通常包括以下几个步骤。
首先,需要获取基因表达数据,比如微阵列或RNA测序数据。
然后,使用适当的算法计算不同基因之间的相关性,常用的方法有Pearson相关系数和Spearman相关系数等。
接下来,选取一定的相关性阈值,将高相关性的基因连接起来形成网络。
最后,通过可视化工具将网络呈现出来,以便进一步分析和解读。
基因共表达网络在生物学研究中有广泛的应用。
首先,它可以帮助我们挖掘基因的功能和调控机制。
通过分析基因共表达网络,我们可以发现潜在的基因相互作用和调控关系。
这些关系可以为我们提供线索,帮助解释基因的功能和参与的生物过程。
其次,基因共表达网络可以帮助我们发现新的基因模块和生物标志物。
模块是在基因共表达网络中高度相互连接的一组基因,这些基因可能组成一个生物功能模块。
通过分析这些模块,我们可以发现新的生物标志物,用于疾病诊断和治疗指导。
此外,基因共表达网络还可用于预测基因功能和疾病相关基因。
在基因共表达网络的分析中,还存在一些挑战和注意事项。
首先,网络构建过程中需要选择合适的相关性阈值。
过低的阈值会导致网络过于复杂和杂乱,而过高的阈值可能会导致网络丢失一些重要的信息。
因此,在阈值选择上需要根据具体研究的问题和数据质量来进行调整。
基因共表达分析基因共表达分析(GeneCo-ExpressionAnalysis)是生物信息学的一个重要的研究手段,可以对基因间的相关关系进行挖掘,并为研究基因表达调控模式提供重要数据支持。
目前,基因共表达分析已经被广泛用于许多生物学领域,如转录组学、基因组学、分子生物学、遗传学和系统生物学等,为相关领域的研究发展提供了重要的技术和理论支持。
一般来说,基因共表达分析是指对一组关联基因的表达水平进行分析,以检测研究基因之间可能存在的关联关系,以及共表达基因及其相关特征。
基于系统生物学方法和测序技术,基因共表达分析可以揭示基因间的信号调控关系,研究基因调控网络,发现功能相关的重要基因,以及基因疾病间的相互关系等。
一般而言,基因共表达分析分为三大类:一是基于非贪婪算法的非监督学习方法,主要是基于数据聚类分析的方法;二是贪婪算法的监督学习方法,通过筛选最具有特征的基因组成的模型,来发现有功能代表性的基因;三是对基因表达数据的直接建模,称为分析模型,旨在发现基因之间较强的共表达关系。
在基因共表达分析中,非贪婪算法的非监督学习方法是用最多的,它具有较好的数据收集和解释性能,从不同角度揭示基因表达、调控、功能和相关性。
主要的非监督学习办法有聚类分析、最近簇分析、主成分分析、层次聚类等。
聚类分析是最常用的非监督基因共表达分析方法,可以根据基因表达水平的相似性将基因分组,确定基因间的关系和共表达规律。
最近簇分析可以确定两个基因表达水平更相近的基因,可以根据这些亲密基因的组合,发现更加复杂的表达模式。
而主成分分析可以将数据降维,从而提取最能够代表数据的主要的特征,帮助我们更加容易地发现基因之间的共表达关系。
最后,层次聚类算法可以根据基因数据的多样性,把基因分组,发现各个基因表达水平变化的趋势。
此外,基因共表达分析还可以与其他常见的数据分析方法相结合,如基于网络的分析方法,以检测基因间的可能联系和共表达关系;还可以结合生物信息学和生物体系学等方法,发现基因互作网络、复杂调控关系等。
遗传关联分析及其在人类疾病研究中的应用遗传关联分析(Genetic Association Analysis)是通过研究基因和表型之间的关系来探讨某个基因是否与某个表型相关的一种方法。
在人类疾病研究中,遗传关联分析是一种重要的工具,它被广泛用于探索人类疾病的遗传基础。
本文将探讨遗传关联分析的基本原理、方法以及在人类疾病研究中的应用。
基本原理遗传关联分析的基本原理是研究基因与表型之间的关系。
由于基因具有复杂性、多样性和多基因性等特点,因此遗传关联分析不仅需要考虑单个基因的影响,而且还需要考虑多个基因之间的相互作用。
遗传关联分析所研究的表型包括所有可能的性状和疾病,例如认知能力、身高、肥胖、糖尿病等。
遗传关联分析的目的是找到影响表型的基因变异,进而探究该基因的生物学功能、作用机制和调控网络等方面的信息。
方法遗传关联分析的方法一般分为两种:关联研究和通路分析。
关联研究是通过比较不同个体(例如患病和健康个体)的基因组序列,在不同群体间比较基因频率的差异来探究某个基因与特定表型的关系。
这种方法可分为两种:候选基因法和基因组广泛关联研究(GWAS)。
候选基因法是基于已知的遗传变异或有理假设的基因,通过比较两个群体的基因频率差异来确定某个基因与表型的相关性。
值得注意的是,这种方法依赖于先前的基因研究。
GWAS是一种在不知道先假设的基因变异的情况下,同时对成千上万的基因进行分析来确定它们与表型之间的关系。
由于GWAS可以无偏地检测大量的基因和基因变异,因此已成为探索人类疾病遗传基础的重要手段。
通路分析是一种系统的方法,它可以探索表型和基因之间的复杂关系。
通路分析通过挖掘基因功能和基因网络之间的交互作用,找到对表型产生重要贡献的基因通路。
在通路分析中,提取有关表型的基因集合,计算这些基因之间的相互作用,并将这些作用转化成一个生物学通路。
这种方法可以帮助科学家深入了解基因和表型之间的关系,并为通过干预基因通路来治疗疾病提供了理论依据。
人工智能在医学教育、科研和临床实践中的应用前景与挑战1. 人工智能在医学教育中的应用前景与挑战随着人工智能技术的快速发展,其在医学领域的应用也日益广泛。
特别是在医学教育方面,人工智能技术为医学生提供了更加高效、个性化的学习方式,同时也为医生的职业发展提供了更多的选择。
在将人工智能技术应用于医学教育的过程中,我们也面临着一些挑战和问题。
人工智能技术在医学教育中的应用需要克服数据不平衡的问题。
医学教育领域的数据往往集中在少数知名高校和研究机构,而广大基层医疗机构和医学院校的数据资源相对匮乏。
这使得人工智能在医学教育中的应用受到限制,无法充分发挥其潜力。
为了解决这一问题,我们需要加强跨机构、跨地区的合作,共享医学教育数据资源,提高数据的覆盖面和质量。
人工智能技术在医学教育中的应用需要关注伦理道德问题,在使用虚拟现实技术进行手术模拟时,如何确保学生的安全以及避免对患者的伤害?如何确保人工智能算法的公平性和透明性,避免加剧社会不平等现象?这些问题需要我们在推动人工智能技术在医学教育中的应用的同时,加强对相关伦理道德问题的探讨和规范。
人工智能技术在医学教育中的应用还需要克服技术瓶颈,尽管人工智能在医学图像识别、自然语言处理等领域取得了显著的成果,但在某些特定场景下,如复杂疾病的诊断和治疗等,人工智能技术仍然存在一定的局限性。
我们需要加大对人工智能技术研发的投入,提高其在医学教育中的实用性和准确性。
人工智能技术在医学教育中具有巨大的应用前景,但同时也面临着诸多挑战和问题。
我们需要在充分认识到这些挑战的基础上,积极探索适合我国国情的人工智能在医学教育中的应用路径,以期为我国医学教育的发展提供有力支持。
1.1 医学影像诊断随着人工智能技术的不断发展,医学影像诊断领域也取得了显著的成果。
人工智能在医学影像诊断中的应用主要包括图像识别、特征提取、分类和预测等方面。
这些技术的应用可以提高医生对疾病的诊断准确性和效率,从而为患者提供更好的医疗服务。
了解生物大数据技术中的基因共表达网络分析流程基因共表达网络分析是生物大数据技术中的一项重要工具,利用这种方法可以揭示基因之间的关联关系,帮助我们理解基因调控网络。
本文将介绍基因共表达网络分析的流程,并解释每个步骤的重要性。
基因共表达网络是基于基因表达模式相似性的网络,其中基因通过它们的表达模式进行连接。
通过共表达网络分析,我们可以识别功能相似的基因模块、预测新基因的功能、发现调控子网络等。
下面是基因共表达网络分析的一般流程:1. 数据获取与预处理:首先,需要从外部数据库或实验室测序得到基因表达数据。
常见的数据来源有公共数据库如GEO、ArrayExpress以及其他测序平台。
数据一般以表达矩阵的形式给出,其中行代表基因,列表示不同样本或条件。
在数据预处理阶段,可以进行质控、标准化、去除离群值等步骤,以保证数据的准确性和可靠性。
2. 相关系数计算:在基因共表达网络中,我们通常使用基因间的相关系数来度量它们的相似性。
常见的相关系数有皮尔逊相关系数、Spearman秩相关系数等。
计算相关系数的目的是根据基因表达数据量化基因间的关联程度。
3. 相关性阈值设定:计算得到的相关系数矩阵会非常庞大,需要设置一个相关性阈值来确定哪些基因之间具有显著的相关性。
相关性阈值可以根据统计方法、经验值或者网络的特性来确定。
设定阈值后,与阈值以下的相关系数将被视为噪音而被舍弃。
4. 网络构建:根据设定的相关性阈值,我们可以构建一个基因共表达网络。
在网络中,基因被表示为网络节点,相关性超过阈值的基因对之间的连接被表示为网络边。
可以使用各种网络构建算法,如连接阈值法、加权关联网络等。
5. 网络分析与可视化:构建完基因共表达网络后,我们可以对其进行进一步的分析和可视化。
常见的网络分析方法包括模块检测、功能富集分析、网络特性分析等。
模块检测是寻找具有相似表达模式的基因子集,功能富集分析可以帮助我们理解这些基因模块的生物学功能,网络特性分析可以量化网络的结构和拓扑特性。
生物信息学中的基因调控网络分析随着高通量测序技术的不断发展,越来越多的基因组学数据被产生和积累。
在这些海量的基因数据中,含有关于基因表达调控的信息。
基因调控是指细胞内的调节机制,控制基因的表达量和表达时机。
这一调控网络是由许多基因和蛋白质相互作用形成的,被称为基因调控网络。
对基因调控网络的分析和理解能够帮助我们深入了解其在细胞生物学和生物信息学中的作用,从而为药物研发、疾病治疗等研究提供理论基础和参考。
基于基因调控网络的分析和挖掘可以用于许多方面。
例如,基因调控网络的分析可以提供关于调控因素的信息,这有助于分析疾病的发病机理及其治疗策略。
此外,基因调控网络的分析还可以帮助预测基因的功能、识别基因表达的生物学过程、评估基因表达的变化等。
基因调控网络分析的常用方法包括基于文献的手工构建和数据驱动的自动构建。
手工构建是指根据文献资料、生物学知识和经验构建基因调控网络。
这种方法虽然精确度较高,但是需要大量的信息搜集和专业知识,无法实现大规模数据的分析。
自动构建是指利用机器学习、统计推断等方法,从基因组学数据中识别和分析基因调控网络。
这种方法可重复性较高,能够承接大量数据处理工作,但是精确度有待提高。
随着人工智能技术的发展,基于深度学习的基因调控网络分析方法也逐渐被开发。
这种方法将基因调控网络看作一个图形结构,使用图卷积神经网络(graph convolutional network)等方法对其进行学习和预测。
深度学习方法有一定的优点,能够处理大规模的基因组学数据,并且可以利用未知的生物学知识对基因调控网络进行预测。
但是,这种方法的模型训练和解释性都需要更进一步研究和提高。
基因调控网络分析的一般流程包括网络构建、网络可视化和功能注释等步骤。
构建基因调控网络需要整合多种数据,如基因表达、转录因子、序列及其亚群、染色体置换和蛋白质-蛋白质相互作用等。
目前已有多种软件和工具可用于构建和分析基因调控网络,其中常用的有Cytoscape、Genome Wide Association Study (GWAS)和Ingenuity Pathway Analysis(IPA)等。
基于文献挖掘和基因共表达网络的基因与疾病关联分析
近年来,高通量技术的发展和研究能力的增长带来了大规模的生物数据。
生物信息学的主要课题之一是从海量数据中挖掘导致疾病的分子机制。
本文基于PubMed文献摘要和由微阵列数据生成的基因共表达网络来挖掘疾病与基因的关联性,并应用于癌症和艾滋病这两种复杂疾病。
随着生物医学相关文献的爆炸性增长,从文献中寻找需要的信息变得越来越困难。
基于关键词的传统搜索引擎难以满足较复杂的搜索需求。
为了解决这一问题,本文提出了语义搜索的广义匹配原则:蕴含检索查询输入语义的目标文本应出现在搜索结果中,并开发
了基于广义匹配原则的语义搜索引擎Sensehit。
Sensehit整合了MeSH、Entrez 基因、UniProt、UnitProt Keywords、基因本体、HGNC、miRBase、HomoloGene等数据库中的生物医学背景知识,基于自然语言处理技术提取PubMed文献摘要中的语义,可用于搜索基因调控模式、蛋白质相互作用、蛋白质修饰、因果关系等生物医学相关信息,为疾病分子机制的研究提供方便。
近年来,许多研究表明microRNA在癌症中扮演着重要的角色。
为了从PubMed文献摘要中寻找和评估microRNA家族与癌症的关联性,本文基于正则表达式识别文本中的microRNA,基于MeSH术语标注获得文献涉及的癌症类型,基于Fisher 精确检验评估microRNA家族和癌症类型的关联性,并建立了记录这
些关联信息的数据库miCancema,可通过Web界面供研究者免费查阅。
miCancerna覆盖的文献数是同类数据库miR2Disease的两倍以上,并达到90%以上的精确度。
同时,本文进一步将其中显著的microRNA与
癌症关联信息构建成关联网络,对该网络的分析表明一些microRNA
家族与特定的癌症类型有关,有可能作为诊断和治疗的靶标,;另一些microRNA家族涉及多种癌症,可能在肿瘤发生中起到关键作用。
导致艾滋病的HIV起源于在非洲一些灵长类动物中传播的SIV。
SIV感染
对天然宿主乌白眉猴无致病性,却会导致非天然宿主恒河猴发展为艾
滋病。
通过对比这两种情况下的基因表达谱,可以探究HIV/SIV感染
导致艾滋病的分子机制。
本文基于乌白眉猴和恒河猴在感染相同SIV 毒株后不同时间点的微阵列基因表达数据,采用皮尔逊相关系数方法
构建了 14个基因共表达网络,对其分析发现,在SIV感染期间,乌白
眉猴和恒河猴基因共表达网络中基因的正、负连接数分布有显著差异。
同时,本文对枢纽基因的信号通路进行了富集分析,得到4个在乌白
眉猴的枢纽基因中显著富集的信号通路,8个在恒河猴的枢纽基因中
富集的信号通路,以及3个在两者的枢纽基因中都富集的信号通路。
进一步分析基因共表达网络中的枢纽基因,可能有助于理解SIV和
HIV感染的发病机制,进而获得预防和治疗艾滋病的新线索。