天然药物化学总结
- 格式:doc
- 大小:2.22 MB
- 文档页数:34

天然药化考试总结Just be happy, remember on the morning of June 18, 2022天然药物化学:是运用现代科学理论与方法;研究天然药物中化学成分的一门学科..研究对象:天然药物中化学成分研究内容:各类天然药物中的结构特点、理化性质、提取分离方法以及主要类型化学成分的结构鉴定等研究的目的及意义:1.阐明天然药物中的药效物质基础;探索中药防治疾病的原理;2.促进药理药效理论研究的深入;3.促进建立和完善中药的质量评价标准;4.扩大药源;为研制开发新药提供理论与实践基础天然药物来源:植物、动物、矿物、微生物和海洋生物一次代谢初生代谢:指对维持植物生命活动必不可少的过程一次代谢物:由一次代谢产生的维持植物生命最基本的;不可缺少的物质二次代谢:在特定的条件下;一些重要的一次代谢产物作为原料或前体;又进一步经历不同的代谢过程二次代谢产物:由二次代谢产生的物质叫做二次代谢产物乙酸—丙二酸途径:通过这一途径可以合成脂肪酸、酚类和蒽酮类化合物甲戊二羟酸途径:萜类和甾体化合物均由这一途径生成桂皮酸及莽草酸途径:具有C6-C3基本骨架的苯丙素、 C6-C3-C6结构的黄酮类化合物均由此途径合成而来氨基酸途径:大多数生物碱类成分由此途径生成复合途径:结构复杂的化合物可由多种生物合成途径复合完成有效成分的提取方法:溶剂提取法、水蒸气蒸馏法、超临界流体萃取法溶剂提取法:煎煮法、浸渍法、渗漉法、回流提取法、连续回流提取法作用原理:溶剂穿透药材粉末的细胞膜;溶解溶质;形成细胞的内外浓度差;将溶质渗出细胞膜;达到提取目的..溶剂的选择相似相溶原理1分类:溶剂按极性大小可以分为三类;即亲脂性溶剂、亲水性溶剂和水.. 2极性:常用溶剂极性由强到弱石油醚<四氯化碳<苯<二氯甲烷<氯仿<乙醚<乙酸乙酯<正丁醇<丙酮<甲醇乙醇<水..3选择要点:根据相似相溶原理;以最大限度提取所需的化学成分;溶剂沸点应适中易回收;低毒、安全..影响溶剂提取的因素:要选择合适的溶剂与方法;对药材的粉碎度、提取温度及时间也有要求;在工业生产中药对这些因素进行优化选择..影响有效成分的提取因素:粉碎度、温度、时间、其他原因煎煮法:将药物粗粉加水加热煮沸提取..特点:方法简便;大部分成分可被不同程度的提取出来..缺点是温度高;对挥发性成分及受热易被破坏的成分不适用..另外;水煎后的药液大多黏度大;滤过困难..浸渍法:将药物粗粉放在容器中;加入水或稀醇浸泡一定时间;反复多次合并浸提液;减压浓缩即可..特点:不用加热;适用于遇热易破坏或挥发性成分;以及粘液质和淀粉较多的成分..但提取时间长;效率低..以水为溶剂时;要防止发霉变质..渗漉法:将药材粗粉装入渗漉筒中;用水或醇作溶剂;先浸渍数小时;然后由下口开始流出提取液;渗漉筒上口不断添加新溶剂;进行渗漉提取..特点:提取效率高于浸渍法..回流提取法:一有机溶剂作为提取溶剂;在回流装置中进行;一般采用反复回流法;即第一次回流一定时间后;滤出提取液;加入新鲜溶剂;重新回流;反复数次合并提取液;减压回收溶剂..特点:效率高于渗漉法;但受热易破坏的成分不宜使用..连续回流提取法:是回流提取法的发展;具有溶剂消耗量小;操作不繁琐;提取效率高的特点..实验室连续回流提取常用索氏提取器或连续回流装置..有效成分的分离纯化方法:溶剂法、沉淀法、分馏法、膜分离法、升华法、结晶法、色谱法溶剂法:酸碱溶剂法、溶剂分配法酸碱溶剂法利用混合物中各组分酸碱性不同而进行分离;溶剂分配法是利用混合物中各组分在两相溶剂中分配系数不同而进行分离沉淀法:专属试剂沉淀法、分级沉淀法、盐析法原理:有的化学成分能与某些试剂生成沉淀;或加入一些试剂后可降低某些成分在溶液中的溶解度而自溶液中析出的一种方法..分馏法:利用混合物中各成分的沸点的不同而进行分离的方法;适用于液体混合物的分离..膜分离法:利用天然或人工合成的高分子膜;以外加压力或化学位差为推动力;对混合物溶液中的化学成分进行分离、分级、提纯和富集..结晶法:利用混合物中各成分在溶剂中的溶解度不同达到分离的方法..溶剂的选择:对被溶解成分的溶解度随温度不同应有显着差别;与被结晶的成分不应产生化学反应;沸点适中;不宜过高、过低色谱法根据分离的原理与机制分类:可分为吸附、分配、凝胶、离子交换色谱等..吸附色谱利用吸附剂对被分离化合物分子的吸附能力的差异而实现分离;凝胶过滤色谱排阻色谱分子筛作用;根据凝胶的孔径和被分离化合物分子的大小而达到分离目的;离子交换色谱离子交换色谱主要靠样品离子与固定相的可交换能力差别而分离;分配色谱利用被分离成分在固定相和流动相之间的分配系数的不同而达到分离;大孔树脂色谱是吸附性和分子筛原理相结合按照操作方式不同可以分为:薄层色谱、柱色谱、纸色谱分配色谱按照流动相和固定相的分子聚集状态分类:气相色谱、液相色谱和超临界流体色谱化合物的纯度测定:薄层色谱、纸色谱、气相色谱或高效液相色谱等化合物的理化鉴定:物理常数的测定、化合物的结构骨架与官能团的确定化合物的波谱测定:质谱、紫外光谱、红外光谱、核磁共振谱、旋光光谱和圆二色光谱结构研究的主要程序:初步推断化合物的类型;测定分子式;计算不饱和度;确定分子中含有的官能团、结构片段或基本骨架;推断并确定分子的平面结构;推断并确定分子的立体结构糖:多羟基醛或多羟基酮及其衍生物、聚合物的总称分子通式为C x H2Oy分类根据分子的大小及其能否水解分类单糖、低聚糖、多聚糖多聚糖:植物多糖淀粉、纤维素、粘液质、果聚糖;菌类多糖猪苓多糖、茯苓多糖、灵芝多糖;动物多糖肝素、透明质酸、硫酸软骨素、甲壳素根据糖的结构特点分类糖匀体、糖杂体糖匀体:单糖、氨基糖、糖醇、去氧糖、糖醛酸糖类与核酸、蛋白质、脂质一起合称生命活动所必需的四大类化合物苷配糖体:糖或糖的衍生物与另一非糖物质通过糖的端基碳原子连接而成的一类化合物非糖部分称为苷元或配基结构:糖的半缩醛羟基与苷元上的羟基脱水缩合;成为具有缩醛结构的物质分类:按苷原子不同分类氧苷、氮苷、硫苷、碳苷氧苷:根据苷键羟基类型不同又分为醇苷、酯苷、酚苷和氰苷..按苷元的化学结构不同分类黄酮苷、蒽醌苷、苯丙素苷、强心苷、吲哚苷等按端基碳构型分类α苷;多为L型;β苷;多为D型按连接单糖个数分类1个糖—单糖苷;2个糖—双糖苷;3个糖—叁糖苷按苷的生理活性分强心苷、皂苷等按生物体内存在状况分原级苷、次级苷按植物来源分人参皂苷、柴胡皂苷等糖和苷的一般性质一性状及溶解度糖:小分子极性大;水溶性好;随着聚合度增高;水溶性下降..多糖难溶于冷水;或溶于热水成胶体溶液.. 单糖极性 > 双糖极性苷:均为固体;糖基少的具有完整的晶形呈结晶状;含糖基多的多为具有吸湿性的无定形粉末..亲水性大小与连接糖的数目、位置有关.. 苷元:为亲脂性(二)苷键的裂解:酸水解、碱水解、乙酰解、酶水解和氧化开裂酸催化水解反应:苷原子先质子化;然后断键生成阳碳离子或半椅式的中间体;中间体再与水结合成糖 从易到难顺序为:N > O > S > C乙酰解反应:与酸催化水解相似;以CH 3CO +即乙酰基;Ac 为进攻基团 常用试剂:醋酐和酸 常用酸:H 2SO 4、HClO 4、CF 3COOH 或Lewis 酸ZnCl 2、BF 3等糖和苷的提取分离(一)提取:主要为溶剂法——水、稀醇单糖、低聚糖、多糖 (二)分离:活性炭柱色谱、纤维素色谱、离子交换柱色谱、凝胶柱色谱、季铵氢氧化物沉淀法、分级沉淀或分级溶解法活性炭柱色谱吸附规律:①对极性基团多的化合物吸附力大于极性基团少的化合物;②对芳香族化合物吸附力大于脂肪族化合物;③对分子量大的化合物吸附力大于分子量小的化合物;④对于糖的吸附力:多糖>低聚糖>单糖 装柱→上样→洗脱顺序为:H 2O 、10%、20%、30%、50%、70%的乙醇液;无机盐、单糖等→二糖→三糖→多糖凝胶柱色谱操作过程:①将凝胶在适当的溶液中浸泡;②待充分膨胀后装入层析柱;③用洗脱液洗脱;④收集、回收溶液;干燥..苷的提取:苯丙素:天然存在的一类含有一个或几个C 6-C 3基团的酚性物质 简单苯丙素:苯丙烯、苯丙酸、香豆素、木脂素等结构与分类:苯丙烯类、苯丙醇类、苯丙醛类、苯丙酸类 提取分离提取:简单苯丙素类一般用有机溶剂或水提取;苯丙烯、苯丙醛及苯丙酸的简单酯类衍生物可用水蒸汽蒸馏法提取;苯丙酸衍生物可用有机酸的常规方法分离:一般可经纤维素、硅胶、大孔树脂或聚酰胺等色谱法分离香豆素类化合物:简单香豆素、呋喃香豆素、吡喃香豆素、其它香豆素类结构:香豆素基本母核;大部分7位有含氧取代分类1、简单香豆素:是指在苯环一侧有取代;且7位OH 未与6或8位取代基2位取代异戊烯基缩合形成呋喃环的香3 4、其它香豆素类:主要包括α-理化性质:石油醚层 氯仿或乙酸乙酯层 苷元、极性小的苷 正丁醇层苷类 水层无机盐、糖、多肽、蛋白质等中药粉末必要时可先脱脂溶剂浸出;回收溶剂O O 12345678(一)荧光:香豆素类在可见光下为无色或浅黄色结晶;在紫外光下可见蓝色荧光(二)内酯的碱水解反应:内酯结构在碱性条件下可水解开环;生成邻羟基桂皮酸盐(三)显色反应1、异羟肟酸铁反应:香豆素具有内酯结构;碱性条件下开环;与盐酸羟胺缩合生成异羟肟酸;在酸性条件下再与Fe3+络合而显红色..2、酚羟基反应:因分子中含有酚羟基所以可以与FeCl3反应产生绿色至黑绿色沉淀..若取代羟基的邻对位无取代可用重氮化试剂反应而显红色至紫红色..3、Gibb’s反应:在PH=9~10时;内酯环水解生成酚羟基;如其对位6位无取代;则与2;6—二氯苯醌亚胺Gibb’s试剂反应而显蓝色..4、Emerson反应:与Gibb’s反应类似;6位无取代时与Emerson试剂4-氨基安替比林和铁氰化钾反应生成红色..提取分离提取:溶剂提取法、碱溶酸沉法、水蒸汽蒸馏法分离:柱色谱、制备薄层色谱和高效液相色谱生物活性:低浓度可刺激植物发芽和生长作用;高浓度则抑制;光敏作用;抗菌、抗病毒作用;平滑肌松弛作用;抗凝血作用;肝毒性木脂素:二苄基丁烷类、二苄基丁内酯类、芳基萘类、四氢呋喃类、双四氢呋喃类、联苯环辛烯类理化性质:木脂素多具有多个手性碳或手性中心;大部分具有光学活性;遇酸易异构化提取分离:溶剂法、碱溶酸沉法、色谱法木脂素的检识常用显色剂:1%茴香醛-浓硫酸试剂;110℃加热5分钟;5%或10%磷钼酸乙醇溶液;120℃加热至斑点出现;10%H2SO4乙醇溶液;110℃加热5分钟;碘蒸气;熏后呈黄棕色;置紫外光下观察荧光..生物活性:抗肿瘤、肝保护和抗氧化作用、中枢神经系统作用、血小板活化因子拮抗作用、抗病毒;抗菌作用、平滑肌解痉作用、毒鱼作用、杀虫作用、其它作用醌类化合物:指具有醌式结构或容易转变为具有醌类性质的化合物;以及在生物合成方面与醌类有密切联系的化合物主要分为苯醌;萘醌、菲醌和蒽醌四类型蒽醌类:一单蒽核类1.蒽醌及其苷类醌母核上常被-OH、-OCH3、-COOH等取代:大黄素型、茜草素型2.蒽酚或蒽酮衍生物(二)双蒽核类:二蒽酮类衍生物、萘骈二蒽酮衍生物理化性质物理性质:升华性及挥发性、不同pH条件下显不同的颜色化学性质∶酸性以游离蒽醌类衍生物为例;酸性强弱将按下列顺序排列:含-COOH > 2个以上β-OH > 1个β-OH > 2个α-OH > 1个α-OH、碱性、颜色反应颜色反应1Feigl反应—醌类氧化还原过程:醌类衍生物在碱性条件下;加热能迅速与醛类及二硝基苯反应;生成紫色化合物2无色亚甲蓝显色试验:在进行薄层色谱检查或纸色谱检查时;无色亚甲蓝作喷雾显色剂;能检出苯醌及萘醌;样品在白色背景上作为蓝色斑点出现3Borntrager’s反应保恩特来格:羟基蒽醌类化合物遇碱显红~紫红色的反应4与活性次甲基试剂反应 Kesting-Craven法:当苯醌及萘醌类化合物的醌环上有未被取代的位置时;即可在氨碱性下与一些含有活性次甲基试剂的醇溶液反应;生成蓝绿色或蓝紫色5与金属离子反应:在蒽醌类化合物结构中;如果有α-酚羟基或具有邻二酚羟基时;则可与Pb2+、Mg2+等金属离子形成络合物;颜色不同;有橙红、紫红、蓝色等6对亚硝基二甲苯胺反应:9或10位未取代的羟基蒽酮类可与0.1%对硝基二甲苯胺吡啶溶液反应产生各种颜色提取分离提取:有机溶剂提取法、碱提取酸沉淀法、水蒸气蒸馏法游离羟基蒽醌的分离:pH梯度萃取法、层析法蒽醌苷的分离:铅盐法、溶剂法生物活性:泻下作用、抗菌作用、扩张冠状动脉的作用、其它作用黄酮类化合物:泛指两个苯环A、B通过三个碳原子相互连接而成C6-C3-C6结构的一系列化合物分类:黄酮类:以2-苯基色原酮为基本母核;3位无含氧取代;B环连接在2位黄酮醇类:基本母核的3位上有含氧取代; B环连接在2位二氢黄酮类:基本母核的2、3位双键被氢化;3位无含氧取代;B 环连在2位二氢黄酮醇类:基本母核的2、3位双键被氢化;3位有含氧取代;B环连接在2位异黄酮类:基本母核为3-苯基色原酮的结构;即B环联结在C环的3位上二氢异黄酮类:基本母核2、3位被氢化;B环连接在3位上查耳酮:氢黄酮C环的1、2位键断裂生成开环衍生物;即三碳链不构成环生物活性:对心血管系统的作用、保护肝脏、抗炎、雌性激素样作用、抗菌及抗病毒、泻下、解痉、抗癌显色反应1.HCl-Mg粉反应:在1ml甲醇或乙醇的样品溶液中;加少许镁粉振摇;再加几滴浓盐酸1~2分钟内可微热即可显色;大多显红→紫红色;少数蓝色或绿色..但查耳酮、橙酮、儿茶素类不显色2.钠汞齐还原反应:在样品的乙醇溶液中加入钠汞齐;放置数分钟至数小时;或加热过滤;绿叶用盐酸酸化则黄酮、二氢黄酮类显红色;黄酮醇类显黄→淡红色;二氢黄酮醇显棕色3.四氢硼钠还原反应:反应后;二氢黄酮醇类被还原产生红→紫红色;其它黄酮类均为负反应4.AlCl3反应:反应生成络合物多呈黄色;在紫外灯下显鲜黄色荧光;但4’-OH黄酮醇或7;4’—二羟基黄酮醇显天蓝色荧光5.锆盐—枸橼酸反应:样品0.5~10mg溶于10ml甲醇;加2%二氯氧锆ZrOCl2甲醇液1ml;则会出现黄色生成了络合物;再加入2%枸橼酸甲醇液;如黄色不退表示有3-OH;如退色表示无3-OH但有5-OH6.氨性氯化锶反应:如黄酮分子中有邻二酚羟基则可与氨性氯化锶反应;产生绿色至棕色乃至黑色沉淀7.三氯化铁反应:黄酮分子中大都含有酚羟基可与FeCl3反应呈现紫、绿、蓝等不同的颜色8.硼酸显色反应:分子中含有下列结构可与硼酸反应产生亮黄色..5-OH 黄酮与6’-OH查耳酮结构符合要求;反应呈阳性;可将这两种与其他区分9.碱性试剂反应:黄酮与碱性试剂可生成黄色、橙色或红色1黄酮类在NaOH水溶液中产生黄~橙色..2查耳酮或橙酮遇碱变红或紫红..二氢黄酮在冷碱中呈黄~橙色;放置或加热变深红~紫红色3黄酮醇碱中先呈黄色;通入空气后由于3-OH氧化;变为棕色..4当分子含3个邻位OH时;在NaOH液中会产生暗绿色或蓝绿色沉淀..提取分离提取:乙醇或甲醇提取法90~95%的醇适于提取游离黄酮;60%左右适于提取黄酮苷、热水提取法仅限于黄酮苷类、碱性水或稀醇提取法分离:溶剂萃取法、PH梯度萃取5%NaHCO3可萃取出7;4’-二羟基黄酮;5%Na2CO3萃取出7或4’-羟基黄酮;0.2%NaOH可萃取出具一般酚羟基的黄酮;4%NaOH萃取出5-OH黄酮、柱色谱法分子中酚羟基越多则吸附越强;形成分子间氢键;难以洗脱;易形成分子内氢键则与聚酰胺的吸附力减小;易被洗脱;分子芳香化程度越高;共轭双键越多;吸附力越强;难以洗脱;不同类型被吸附强弱顺序为:黄酮醇>黄酮>二氢黄酮>异黄酮;若洗脱剂为含水系统时;黄酮苷比游离态先洗脱下来萜类化合物:凡由甲戊二羟酸衍生、且分子式符合C5H8n 通式的衍生物物合成:1.经验异戊二烯法则萜类化合物均由异戊二烯单位以头尾顺序或非头尾顺序相连而成;生源异戊二烯法则萜类化合物由甲戊二羟酸衍生而成提取分离提取:1、溶剂提取法:对于非苷性化合物用甲醇或乙醇提取;回收溶剂后加水悬浮;再用乙酸乙酯萃取;也可用不同极性溶剂依次萃取..苷类可先用乙醚或石油醚萃取;除去强脂溶性杂质;水溶液再用正丁醇萃取;回收正丁醇即得总苷..2、碱提酸沉法:适用于含内酯环的萜类;在热碱中开环成盐溶于水;酸化后又闭环析出;但对酸碱易引起结构发生不可逆变化的萜内酯不可用该法..3、吸附法:利用活性炭和大孔树脂吸附水溶液中的萜苷后;先用水及稀醇依次洗去水溶性杂质;再用合适浓度的乙醇洗脱萜苷..例如:桃叶珊瑚苷及甜叶菊苷可分别用活性炭和大孔树脂纯化获得..分离:结晶法、利用特殊官能团分离、柱色谱常用吸附剂:硅胶、中型氧化铝;洗脱剂:石油醚、正己烷、环己烷、苯等单一溶剂加入不同比例的乙酸乙酯或用氯仿-甲醇洗脱挥发油的提取1.蒸馏法:是最常用的方法;可以共水蒸馏、隔水蒸馏或水蒸气蒸馏2.溶剂法:用低沸点的有机溶剂回流提取或冷浸3.吸收法:用于贵重的挥发油如玫瑰油;茉莉花油等4.压榨法:用于含量多的原料如新鲜的橘子皮、柠檬皮等..5.超临界二氧化碳流体提取;可防止受热不稳定的挥发油;可提高品质6.冷冻析晶法:将挥发油于0℃以下放置使结晶析出分离7.分馏法:挥发油的组成成分类别不同;沸点有差别;可用分馏法大致分离不纯需纯化..8.化学分离法:碱性成分的分离;酚、酸性成分的分离;醇类的分离;醛、酮的分离9.色谱分离法:吸附柱色谱最常用硅胶和氧化铝柱色谱..洗脱剂多用石油醚或己烷;混以不同比例的乙酸乙酯三萜类化合物:由30个碳原子组成的萜类化合物;分子中有6个异戊二烯单位..组成:由苷元四环或者五环三萜和糖组成..糖链:单糖链、双糖链、三糖链成苷位置:3-OH、28-COOH酯苷、其它位-OH原生皂苷:天然产生;未发生水解的皂苷次生皂苷:原生苷的糖被部分降解的产物四环三萜:达玛烷型C位有角甲基且为β构型、羊毛脂烷型3S-环氧鲨烯8通过椅-船-椅式构象形成、甘遂烷型、环阿屯烷型、葫芦烷型、楝烷人参总皂苷不能表现出溶血的现象达玛烷型的人参皂苷在HCl溶液中加热煮沸水解;会发生差向异构化只能得到人参二醇和人参三醇;得不到原生皂苷元原人参二醇和原人参三醇..若要得到原人参皂苷元;必须在缓和的条件下水解20S-原人参三醇衍生的皂苷有溶血性质;而由20S-原人参二醇衍生的皂苷具对抗溶血的作用;因此人参总皂苷不能表现出溶血的现象..五环三萜:齐墩果烷型、乌苏烷型、羽扇豆烷型、木栓烷型三萜类化合物的理化性质溶解度:苷元多有较好结晶;能溶于石油醚、苯、乙醚、氯仿等有机溶剂;而不溶于水;成苷后;极性加大;不易结晶;多为无色定形粉末;可溶于水;易溶于热水;醇中;难溶于乙醚、苯等极性小的有机溶剂;含水丁醇或戊醇对皂苷的溶解度较好..皂苷多数具有苦而辛辣味;吸入鼻内能引起喷嚏..颜色反应1醋酐-浓硫酸反应:可产生黄-红-紫-蓝等颜色最后褪色..2五氯化锑反应:SbCl5显灰蓝色、灰紫色等多种颜色..3三氯醋酸反应:生成红色-蓝色..4氯仿-浓硫酸反应:硫酸层呈现红色或蓝色;氯仿层有绿色荧光出现..5冰醋酸-乙酰氯反应:淡红色或紫红色..表面活性溶血作用沉淀反应:皂苷水溶液可和金属盐如铅盐、钡盐、铜盐等产生沉淀三萜皂苷元提取:1、醇类溶剂提取后;提取物依次用石油醚、氯仿、乙酸乙酯等溶剂进行分部提取;然后进一步分离 ;三萜苷元主要来自氯仿部位;2、以皂苷形式存在的;水解后用氯仿等溶剂萃取;然后进行分离三萜皂苷提取:用稀醇提取;提取液减压浓缩后;加适量水;必要时先用石油醚等萃取;去杂;后用正丁醇萃取;减压蒸干;通过大孔吸附树脂;水洗去糖等;后用30%~80%甲醇或乙醇梯度洗脱;洗脱液减压蒸干;得粗制总皂苷..用重结晶、层析等方法分离纯化皂苷..三萜皂苷分离:分段沉淀法:由于皂苷难溶于乙醚、丙酮等溶剂;故可将粗皂苷先溶于少量乙醇中;然后逐渐加入乙醚、丙酮、皂苷即可析出;可逐渐降低溶剂极性;不同极性的皂苷就会分批沉淀出来;但效果不太好..吸附柱色谱:正相或反相硅胶柱色谱、分配柱色谱、高效液相色谱、大孔树脂正相硅胶色谱:常用硅胶;用混合有机溶剂洗脱;反相硅胶色谱:常用Rp-18、Rp-8反相硅胶;用甲醇-水混合系统洗脱..分配柱色谱:由于皂苷极性大;常用硅胶作支持剂;3%草酸水溶液为固定相;流动相为含水的混合有机溶剂高效液相色谱:分离效能较高;采用反相色谱柱;以甲醇-水、乙腈-水等系统洗脱大孔树脂:分离极性较大的化合物;尤其适用于皂苷的精制和初步分离;将含有皂苷的水溶液上大孔树脂柱后;先用水洗涤除去糖和其他水溶性杂质;然后再用不同浓度的甲醇和乙醇依照浓度由低到高的顺序进行梯度洗脱..甾体化合物类固醇化合物:化学结构中具有甾体母核----环戊烷骈多氢菲的一类化合物主要包括:植物甾醇、胆汁酸、C21甾类侧链为羟甲基衍生物、昆虫的变态激素、强心苷侧链为不饱和内酯环、甾体皂苷侧链为含氧螺杂环、甾体生物碱、蟾毒配基等颜色反应1.醋酐-浓硫酸:样品溶于冰醋酸;加浓硫酸-醋酐1:20;产生红红→紫→蓝→绿→污绿等颜色变化;最后褪色..2.Salkowski氯仿-浓硫酸反应:样品溶于氯仿;沿管壁滴加浓硫酸;氯仿层显血红色或青色;硫酸层显绿色荧光..3.三氯乙酸反应:与25%三氯乙酸乙醇溶液反应呈红→紫色..。

第一章总论天然药物化学是运用现代科学理论与方法研究天然药物中化学成分的一门学科;其研究内容包括各类天然药物的化学成分主要是生理活性成分或药效成分的结构特点、物理化学性质、提取分离方法以及主要类型化学成分的结构鉴定等;一.中草药有效成分的提取从药材中提取天然活性成分的方法有溶剂法、水蒸气蒸馏法及升华法等;●溶剂提取法的原理:溶剂提取法是根据“相似相容”原理进行的,通过选择适当溶剂将中药中的化学成分从药材中提取出来的一种方法;考试时请这样回答哦常用溶剂极性有弱到强排列:石油醚<环己烷<苯<乙醚<氯仿<醋酸乙酯<正丁醇<丙酮<乙醇<甲醇<水丙酮,乙醇,甲醇能够和水任意比例混合;常用溶剂的性质:亲脂性有机溶剂、亲水性有机溶剂、水一般情况下,分子较小,结构中极性基团较多的物质亲水性较强;而分子较大,结构上极性基团少的物质则亲脂性较强;●天然药物中各类成分的极性·多糖、氨基酸等成分极性较大,易溶于水及含水醇中;·鞣质是多羟基衍生物,列为亲水性化合物;·苷类的分子中结合有糖分子,羟基数目多,能表现强亲水性;·生物碱盐,能够离子化,加大了极性,就变成了亲水性化合物;·萜类、甾体等脂环类及芳香类化合物因为极性较小,易溶于氯仿、乙醚等亲脂性溶剂中;·油脂、挥发油、蜡、脂溶性色素都是强亲脂性成分,易溶于石油醚等强亲脂性溶剂中总之,天然化合物在溶剂中的溶解遵循“相似相溶”规律;即极性化合物易溶于极性溶剂,非极性化合物易溶于非极性溶剂,分子量太大的化合物往往不溶于任何溶剂;溶剂提取法的关键是选择适宜的溶剂选择溶剂依据:根据溶剂的极性和被提取成分及其共存杂质的性质,决定选择何种溶剂各溶剂法分类见天然药物化学辅导教材P5三水蒸气蒸馏法只适用于具有挥发性、能随水蒸气蒸馏而不被破坏,与水不发生反应,且难溶或不溶于水的成分的提取;天然药物中的挥发油、某些小分子生物碱如麻黄碱、烟碱、槟榔碱以及某些小分子的酚性物质如牡丹酚等的提取可采用水蒸气蒸馏法;四升华法某些固体物质如水杨酸、苯甲酸、樟脑等受热在低于其熔点的温度下,不经过熔化就可直接转化为蒸气,蒸气遇冷后又凝结成固体称为升华;天然药物中有一些成分具有升华性质,能利用升华法直接中药材中提取出来;但天然药物成分一般可升华的很少;果蔬脱水新技术实质上升华脱水法;五超临界二氧化碳流体萃取法了解部分,见天然药物化学辅导教材P6三、中草药有效成分的分离与精制一根据物质溶解度不同进行分离1. 原理: 相似相溶2. 方法: 结晶法、试剂沉淀法、酸碱沉淀法、铅盐沉淀法、盐析法二根据物质分配系数的不同进行分离K = CU / CLCU:上相,CL:下相,K值与萃取次数成反比,即K值越大,萃取次数越少,反之越多;⑴分配系数K值与萃取次数的关系原理: 利用物质在两种互不相溶的溶剂中的分配系数的不同达到分离 ;分配系数K值:一种溶质在两相溶剂中的分配比;K值在一定的温度和压力下为一常数;⑵分离因子β值与分离难易的关系分离因子β:两种溶质在同一溶剂系统中分配系数的比值;b = KA / KB KA>KBb值越大,越易分离; b =1时,无法分离;⑶酸碱度pH值对分配比的影响溶剂系统PH的变化影响酸性、碱性、及两性有机化合物的存在状态游离型或离解型,从而影响在溶剂系统中的分配比;游离型------极性小的溶剂;离解型-------极性大的溶剂◆PH<3,酸性物质多呈游离型HA、碱性物质则呈离解型BH+;◆ PH>12,酸性物质呈离解型A-、碱性物质以游离型B存在;纸色谱法 PC以滤纸纤维为惰性载体的平面色谱支持剂:纤维素滤纸固定相:纤维素上吸附的水20-25%展开剂:与水不相混溶的有机溶剂或水饱和的有机溶剂Rf值: A、物质极性大, Rf值小; B、物质极性小, Rf值大;应用:适合于分离亲水性较强的物质;液-液分配柱色谱法固定相主要为化学键合柱色谱:将吸附固定液的载体装入色谱管中进行分离和检测混合物成分的色谱法;按是否加压分:常压柱色谱、加压柱色谱按相极性分:正相色谱、反相色谱载体:硅胶含水17%以上、硅藻土及纤维素等●正相色谱:固定相>流动相极性固定相:水、缓冲溶液流动相:氯仿、乙酸乙酯、丁醇等弱极性有机溶剂洗脱顺序:极性小的化合物先出柱,极性大的化合物后出柱应用:适用于水溶性或极性较大的化合物,如生物碱、苷、糖类、有机酸等;●反相色谱:固定相<流动相极性固定相:石蜡油,化学键合相如十八烷基硅胶键合相流动相:水、甲醇、乙腈等强极性有机溶剂洗脱顺序:极性大化合物,先出柱;极性小化合物,后出柱;应用:适合于脂溶性成分,如高级脂肪酸、油脂、游离甾体等;(三)..根据物质吸附性差别进行分离吸附色谱法利用同一吸附剂对混合物中各成分吸附能力的不同而达到分离的色谱方法;吸附类型:1.物理吸附溶液分子与吸附剂表面分子的分子间作用力:硅胶、氧化铝及活性炭为吸附剂的吸附;相似者易吸附2.化学吸附:如黄酮等酚酸性物质被碱性氧化铝吸附,生物碱被酸性硅胶吸附等;3.半化学吸附:如聚酰胺与黄酮类、蒽醌类等化合物之间的氢键吸附;介于物理吸附与化学吸附之间;固-液吸附柱色谱将待分离混合物样品加在装有吸附剂的柱子中,再加适当的溶剂洗脱剂冲洗,由于吸附剂对各组分吸附能力不同,各组分在柱中向下移动的速度不同,吸附力最弱的组分随溶剂首先流出,通过分段定量收集洗脱液而使各组分得到分离;固-液吸附三要素:吸附剂、溶质、溶剂●吸附剂的种类及特点 1.极性吸附剂氧化铝、硅胶特点:a.对极性强的物质吸附能力强;b.溶剂极性减弱,则吸附剂对溶质的吸附能力增强;反之,则减弱;c.溶质即使被硅胶、氧化铝吸附,一旦加入极性较强的溶剂时,又可被置换洗脱下来;为避免化学吸附,酸性物质宜用硅胶、碱性物质宜用氧化铝作为吸附剂进行分离;通常在分离酸性或碱性物质时,洗脱溶剂中常加入适量的醋酸或氨、吡啶、二乙胺,以防止拖尾,改善分离效果;●非极性吸附剂活性炭特点:活性炭因为是非极性吸附剂,对非极性物质具有较强的亲和能力;在水中对溶质表现出强的吸附能力,溶剂极性降低,则活性炭对溶质的吸附能力也随之降低;故从活性炭上洗脱被吸附物质时,洗脱溶剂的洗脱能力将随溶剂极性的降低而增强;当用活性炭作吸附剂进行层析时,下列洗脱剂的洗脱能力由小列大为:水、l0%、20%、30%、50%、75%、95%的乙醇;聚酰胺吸附色谱法通过分子中的酰胺羰基与酚类、黄酮类化合物的酚羟基,或酰胺键上的游离胺基与醌类、脂肪酸上的羰基形成氢键缔合而产生吸附;●吸附强弱规律含水溶剂中a.形成氢键的基团数目越多,则吸附能力越强;形成氢键的能力与溶剂有关,一般在水中形成氢键的能力最强,在有机溶剂中较弱,在碱性溶液中最弱;c.分子中芳香化程度越高,则吸附性能越强;b.易形成分子内氢键的化合物,其吸附性能减弱;在聚酰胺柱色谱分离时,通常用水装柱,样品也尽可能作成水溶液上柱以利聚酰胺对溶质的充分吸附,形成较窄的原始谱带;随后用不同浓度的含水醇洗脱,并不断提高醇的浓度,逐步增强从柱上洗脱物质的能力;甲酰胺、二甲基甲酰胺及尿素水溶液因分子中均有酰胺基,作为第三者可以同时与聚酰胺及酚类等化合物形成氢键缔合,故有很强的洗脱能力;此外,水溶液中加入碱或酸均可破坏聚酰胺与溶质之间的氢键缔合,也有较强的洗脱能力;●各种溶剂在聚酰胺柱上的洗脱能力由弱至强排序为:水→甲醇→丙酮→氢氧化钠水溶液→甲酰胺→二甲基甲酰胺→尿素水溶液●应用 a.特别适合于酚类、醌类、黄酮类化合物的制备和分离;b.对生物碱、萜类、甾体、糖类、氨基酸等其它极性与非极性化合物的分离也有着广泛应用;c.用于提取物的脱鞣质处理大孔吸附树脂的吸附由于吸附性和分子筛原理,有机化合物吸附力的不同及分子量的不同,在大孔吸附脂上经一定的溶剂洗脱而分开; ①吸附性-----范德华引力或产生氢键的结果;②分子筛------本身多孔性结构所决定; 大孔吸附树脂:分为极性和非极性●影响因素:a.一般非极性化合物在水中易被非极性树脂吸附,极性化合物易被极性树脂吸附;糖是极性的水溶性化合物,与D型非极性树脂吸附作用很弱,据此经常用大孔吸附树脂将中药的化学成分和糖分离;b.物质在溶剂中的溶解度大,树脂对此物质的吸附力就小,反之就大;c.分子量小、极性小的化合物与非极性大孔吸附树脂吸附作用强;反之,与极性大孔吸附树脂吸附作用强;d.能与大孔吸附树脂形成氢键的化合物易吸附;●洗脱液的选择:最常用的水、乙醇、甲醇、丙酮、乙酸乙酯对非极性大孔树脂:洗脱液极性越小,洗脱能力越强;对极性大孔树脂:洗脱液极性越大,洗脱能力越强;●应用广泛应用于天然化合物如苷与糖类的分离、生物碱精制;主要用于水溶性大分子化合物的分离和精制:如多糖、蛋白质、多肽类化合物分离;四根据物质分子大小差别进行分离凝胶色谱法:将含有大小不同分子的混合物样品液,通过多孔性凝胶固定相,用洗脱剂将分子量由大到小的化合物先后洗脱的一种分离方法;五根据物质解离程度不同进行分离天然有机化合物中,具有酸性、碱性及两性基团的分子,在水中多呈离解状态,据此可用离子交换法或电泳技术进行分离;以下仅简单介绍离子交换法;●.原理:是以离子交换树脂作为固定相,用水或含水溶剂为流动相;当流动相流过交换柱时,溶液中的中性分子及不与离子交换树脂交换基团发生交换的化合物将通过柱子从柱底流出,而具有可交换的离子则与树脂上的交换基团进行离子交换并被吸附到柱上,随后改变条件,并用适当溶剂从柱上洗脱下来,即可实现物质分离;●结构及性质:离子交换树脂外观均为球形颗粒,不溶于水,但可在水中膨胀;●吸附规律:阳离子交换树脂——分离碱性成分;阴离子交换树脂——分离酸性成分●.应用:主要用于能产生离子型的成分如氨基酸、肽类、生物碱、有机酸、酚类等;四、结构研究法结构测定常用的波谱分析紫外-可见吸收光谱uv凡具有不饱和键的化合物,特别是存在共扼不饱和键的化合物,在紫外-可见光谱200-700 nm中有特征吸收峰,所以紫外光谱适用于鉴定不饱和键的有无,或用以推测这些不饱和键是否共扼; 红外光谱 IR红外光谱能充分反映官能团与波长的关系,所以对确定未知物的结构非常有用; 常见官能团伸缩振动区:①O-H、N-H 3750-3000 cm-1 ②C-H 3300-2700 cm-1③C≡C2400-2100 cm-1 ④C=O 1900-1650 cm-1 ⑤C=C 1690-1600 cm-1质谱 MS就是化合物分子经电子流冲击或用其他手段打掉一个电子后,形成正电离子,在电场和磁场的作用下,按质量大小排列而成的图谱;用质谱测定有机分子的分子量;核磁共振谱NMR1H–NMR和13C-NMR能提供分子中有关氢及碳原子的类型、数目、互相连接方式、周围化学环境以及构型、构象等结构信息;●氢谱H—NMR1H –NMR通过测定化学位移δ、质子数以及裂分情况重峰数及偶合常数J可以得出分子中1H 的类型、数目及相邻原子或原子团的信息;①化学位移:在有机化合物中虽同为氢核,如果它们所处的化学环境不同,则它们共振时所吸收的能量就稍有不同,在波谱上就显示出共振峰位置的移动;这种因化学环境变化引起的共振谱线的位移称为化学位移,用符号δ表示;②质子数:根据氢谱的上峰的积分面积并结合已知的分子式求得每个信号所相当的氢的个数,现在1H–NMR 可以直接给出每个信号代表的质子的个数,并可以直接获得分子中总的质子数;③信号的裂分及偶合常数J:磁不等同的两个或两组1H核在一定距离内会因相互自旋偶合干扰而使信号发生裂分,而出现ssinglet,单峰、ddoublet,双峰、ttriplet,三重峰、uartet,四重峰、mmultiplet,多重峰等;峰裂分数:n+1规律④裂分间的距离称为偶合常数J,单位Hz;其大小取决于间隔键的距离;间隔的键数越少,则J的绝对值越大;反之,越小;按间隔键的多少可分为偕偶J2 、邻偶J3及远程偶合J远 ;※一般相互偶合的两个或两组1H核信号其偶合常数相等Jab=Jba;课后作业一、名词解释1.天然药物化学:是指运用现代科学理论与方法研究天然药物中化学成分的一门学科;其学习内容包括天然药物化学的化学成分的结构特点、物理化学性质、提取分离以及主要类型化学成分的结构鉴定等等;2.有效成分:是指具有生理活性有药效、能治病的成分; 有效部位:是指具有一种主要有效成分或组成相似的有效成分的部位;无效成分:没有生理活性、没有药效、不能治病的成分4.溶剂提取法、系统溶剂提取法略第二章糖和苷概述糖是多羟基醛或酮类化合物及其聚合物;苷的共性是糖和苷键;第一节单糖的立体化学一、单糖结构式的表示方法:优势构象式、Haworth、FischerFischer投影式⑴主碳链上下排列,取代基左右排列;⑵羰基一端在上方;⑶主碳链上下两端价键和所结合的基团指向纸面后方,水平方向的价键和与之相结合的基团指向纸面前方;※因此,Fischer投影式只能在纸面上转动n180n=1,2,3…或转n90°,而不能使之翻转二、单糖的氧环各种糖之间的转化三、单糖的绝对构型Fischer投影式:看距羰基最远的不对称C-OH,OH向右———D型; OH向左———L型;Haworth投影式:看不对称C-R的朝向旋转R面上———D型; R面下———L型;四、单糖的端基差向异构单糖成环后形成了一个新的手性碳原子,该碳原子为端基碳,形成一对异构体为端基差向异构体,有α、β两种构型;Fischer投影式:看距羰基最远的不对称C-OH与C1-OH关系同侧——α型异侧——β型;Haworth投影式:看距羰基最远不对称C-R与C1-OH关系旋转异侧———α型;同侧———β型;五、单糖的构象呋喃糖的五元氧环基本为一平面;吡喃糖的六元氧环有船式和椅式两种构象,以椅式C为主;根据C椅式的存在形式又可分为C1式和1C式;直立键和平伏键的具体写法:①在C1式中位于C4、C2面上和C1、C3、 C5面下的基团为竖键;②平伏键e键与环上的键隔键平行; ③横键或竖键在环的面上面下交替排列;·α-L、β-D ,C1式 ,C1-OH在e键平伏键·α-D、β-L ,C1式 ,C1-OH在a键直立键第二节糖和苷的分类糖类物质根据其能否水解和分子量的大小分为单糖、低聚糖、多糖一.单糖类天然单糖以五碳糖、六碳糖最多,多数在生物体内呈结合状态,只有葡萄糖、果糖等少数以单糖存在;结构见课本p57二.低聚糖由2-9个单糖通过苷键结合而成的直链或支链聚糖称为低聚糖;·按单糖个数可以分为二糖、三糖等·按是否具有还原性分为还原糖和非还原糖·具有游离醛基或通基的糖为还原糖;如果二糖都以半缩醛或半缩酮上的羟基通过脱水缩合而成的聚糖没有还原性,为非还原糖;三、多聚糖由十个以上的单糖通过苷键连接而成的糖;①植物多糖:淀粉、纤维素、果聚糖、半纤维素、树胶、粘液质②动物多糖:糖原、甲壳素、肝素、硫酸软骨素、透明质酸四、苷类苷是由糖及其衍生物的半缩醛或半缩酮的羟基与非糖物质苷元脱水形成的一类化合物;新生成的化学键即位苷键;知道各类特点即可第三节糖和苷的性质一、糖和苷的物理性质●溶解性糖:小分子极性大,水溶性好,随着聚合度增高,水溶性下降;多糖难溶于冷水,或溶于热水成胶体溶液,难溶于高浓度的乙醇;单糖极性 > 双糖极性 ;①苷——亲水性其大小与连接糖的数目、性质有关;※ C-苷在水或有机溶剂中的溶解度都较小;②苷元——为亲脂性; 可溶于乙醚、氯仿等有机溶剂中;●味觉①单糖~低聚糖——甜味; ②多糖——无甜味;随着糖的聚合度增高,则甜味减小;③苷类——苦人参皂苷、甜甜菊苷等;●旋光性:数值上相接近的一个便是与之有相同苷键的一个;利用旋光性→测定苷键构型※糖有旋光性;天然存在的单糖左旋、右旋的均有,但以右旋的较多;※苷类具有旋光性,天然苷类多呈左旋;苷类水解后,由于生成的糖常是右旋的,因而使混合物呈右旋;二、糖和苷的化学性质●氧化反应:单糖分子中有醛酮、伯醇、仲醇和邻二醇等结构①其易氧化程度为:醛酮基>伯醇基>仲醇基 . ②反应速度:顺式>反式因顺式易形成环式中间体.③对固定在环的异边并无扭曲余地的邻二醇羟基不反应;④.反应在水溶液中进行或含水溶液;⑤反应定量进行;●糠醛酚醛缩合反应;也叫Molish反应-----是糖的检识反应,也是苷类的检识反应;现象:界面处紫色环; ※碳苷和糖醛酸与Molish试剂往往不反应;第四节苷键的裂解1、按裂解的程度可分:全裂解和部分裂解;2、按所用的方法可分:均相水解和双相水解;3、按照所用催化剂的不同可分:酸催化水解、碱催化水解、酶解、过碘酸裂解、乙酰解等;●酸催化水解:阳碳离子酸水解难易程度规律有利于苷键原子质子化和中间体形成的因素均有利于水解;①按苷键原子的不同,苷类水解从易到难的顺序为:N-苷> O-苷> S-苷> C-苷;注意:N碱性最强,最易质子化,所以N-苷最易水解;②N-苷的N原子在酰氨及嘧啶环上,很难水解由于受到强的吸电子效应,碱性几乎消失;③酚苷及烯醇苷比其它醇苷易水解;如苯酚苷因苷元部分有供电结构;④.2,6-二去氧糖苷>2-去氧糖苷>6-去氧糖苷>2-羟基糖苷>2-氨基糖苷由于氨基、羟基均可与苷键原子争夺质子⑤呋喃糖苷>吡喃糖苷因五元呋喃环中各取代基处在重叠位置,水解时形成中间体使张力减小;酮糖多为呋喃糖结构,醛糖多为毗喃糖结构,故酮糖苷较醛糖苷易水解;⑥.在吡喃糖苷中由于C5-R会对质子进攻苷键造成一定的位阻,故R愈大,则愈难水解;五碳糖苷>甲基五碳糖苷>六碳糖苷>七碳糖苷>糖醛酸苷⑦当苷元为小基团——横键的苷键比竖键易水解,横键上原子易于质子化当苷元为大基团——苷键竖键比横键易水解;苷的不稳定性促使其水解●碱催化水解通常苷键对碱稳定,但某些特殊的苷如:酯苷、酚苷、与羰基共轭烯醇苷——易被碱水解●酶催化水解反应反应条件温和、专属性高、能够获得原苷元常用的苷键水解酶:杏仁苷酶—水解—β-六碳醛糖苷键纤维素酶—水解—β-D-葡萄糖苷键麦芽糖酶—水解—α-D-葡萄糖苷键转化糖酶—水解—β-果糖苷键●过碘酸裂解反应Smith降解法·特点:反应条件温和、易得到原苷元;可通过产物推测糖的种类、糖与糖的连接方式以及氧环大小;·适用范围:苷元不稳定的苷和碳苷得到连有一个醛基的苷元,不适合苷元上有邻二醇羟基或易被氧化的基团的苷;·所用试剂为:NaIO4、NaBH4·产物:多元醇、羟基乙醛、苷元·碳苷是很难用酸催化水解的,而用Smith裂解获得连有一个醛基的苷元;第五节糖及苷的提取分离一、提取▲糖苷类具多羟基,极性较大,易溶于水,难溶于低极性有机溶剂,但苷类化合物的溶解度则因苷元性质不同而有较大差异;▲糖的提取方法:根据它们对水和醇的溶解度不同而采用不同的方法;如单糖包括小分子低聚糖可用水或50 %醇提取;多糖根据可溶于热水,而不溶于醇的性质提取;依据:①多糖溶于热水中,采用水煎煮法提取;②多糖不溶于醇,采用逐步提高醇的浓度、使多糖分级在醇中析出,以达到纯化和分离;▲苷类提取常用的方法:※若提取的是原生苷,需抑制或破坏酶的活性,采用热乙醇或沸水提取;※若提取次生苷可用酶解方法,酶解后用适当浓度醇或乙酸乙酯提取;※若提取苷元可先酸水解或酶解,再用低极性有机溶剂乙醚或氯仿提取;抑制或破坏酶活性的方法:①在中药中加入一定量的碳酸钙②采用甲醇、乙醇或沸水提取③在提取过程中还须尽量勿与酸和碱接触;否则,得到的不是原生苷,而是已水解失去一部分糖的次生苷,甚至是苷元;二、分离●活性炭柱层析:活性碳为非极性吸附剂,吸附量大、分离率高;对于糖的吸附力:多糖 > 低聚糖 > 单糖方法以活性碳装柱→上样→水洗脱单糖→递增浓度乙醇洗脱二糖、三糖、低聚糖、直至总苷被依次洗脱;●凝胶柱层析:利用分子筛原理;对于不同聚合度的糖类及其水溶性成分的分离特别有效,方法快速、简单、条件温和;洗脱顺序:随分子量由大及小依次流出;●离子交换柱色谱①除去水提液中的酸、碱性成分和无机离子;②制成硼酸络合物——强碱性阴离子交换树脂不同浓度的硼酸盐洗脱●季铵盐沉淀法●.分级沉淀法●蛋白质去除法三、糖和苷的检识利用糖的还原性和糖的脱水反应所产生的颜色变化、沉淀生成等现象来进行理化检识,利用纸色谱和薄层色谱进行色谱检识;●理化检识①.Molish反应:检识糖或苷类化合物;若在两液面间有紫色环产生,则含有糖或苷类化合物;②.Fehling试剂反应:检验还原糖存在;③.Tollen反应:检验还原糖存在;●色谱检识★纸色谱 PC ★薄层色谱 TLC比较下列成分苷元相同Rf值的大小:苷元<单糖苷<双糖苷特点:增加糖在固定相中溶解度,使硅胶吸附能力下降,利于斑点集中,可增加样品载样量;显色剂:除纸层析外,还有—硫酸/乙醇液、茴香醛-硫酸试剂、苯胺-二苯胺磷酸试剂;思考:1.写出Smith裂解反应的反应式;2.写出D-葡萄糖、L-鼠李糖、D-葡萄糖醛酸、芸香糖的结构式;3.苷键具有什么性质,常用哪些方法裂解苷类的酸催化水解与哪些因素有关水解难易有什么规律4.苷键的酶催化水解有什么特点;第三章苯丙素类概述:苯丙素是一类含有一个或几个C6-C3单位的天然成分;第一节苯丙酸类结构特点: C6-C3结构,具有酚羟基取代的芳香羧酸;熟悉常见苯丙酸类型结构:对羟基桂皮酸、咖啡酸、阿魏酸、芥子酸;第二节香豆素类是顺邻羟基桂皮酸的内酯,具有芳香气味;其基本骨架为苯骈α-吡喃酮,7-位常有羟基或醚基;部分香豆素在生物体内以邻羟基桂皮酸苷的形式存在,酶解后苷元邻羟基桂皮酸立即内酯化而成香豆素;一、香豆素的结构类型●简单香豆素类七叶内酯只在苯环上有取代的香豆素类;取代基包括羟基、甲氧基、亚甲二氧基和异戊烯氧基等;多数在7位上有含氧官能团的存在;异戊烯氧基除直接在O上外,在6和8位出现多电负性高●呋喃香豆素类---环合时脱去3个C 苯环上的异戊烯基与邻位酚羟基环合成呋喃环 ;①线型6 ,7呋喃骈香豆素型:C6-异戊烯基和C7-OH环合补骨脂内酯。

天然药物化学期末知识点整理一、天然药物化学成分的分类与结构特点1、糖类:单糖、低聚糖、多糖2、氨基酸和蛋白质3、脂类和蜡类4、色素类5、醚类、醛类与酮类6、苯丙素类7、醌类8、黄酮类9、萜类10、挥发油11、有机酸类12、酚酸类13、苯丙酸类14、其他类二、天然药物化学成分的提取与分离方法1、溶剂提取法2、水蒸气蒸馏法3、升华法4、超临界流体萃取法5、膜分离技术6、色谱技术7、波谱技术(红外、紫外、核磁、质谱)三、天然药物化学成分的结构鉴定方法1、紫外光谱法2、红外光谱法3、质谱法4、核磁共振谱法5、X射线衍射法6、计算机模拟方法7、其他方法(化学方法、色谱-波谱联用技术)四、天然药物化学成分的理化性质与稳定性1、物理性质(溶解度、旋光性、熔点、沸点等)2、化学性质(酸碱性、氧化还原性、水解性等)3、稳定性(温度、湿度、光照等影响因素)4、生物活性与药理作用机制5、安全性评价与质量控制标准6、临床应用与常见剂型及用法用量7、中药复方的配伍规律与作用机制8、新药研发过程中的天然药物化学研究策略与思路在即将到来的期末考试之前,对经济学基础课程的重要知识点进行系统性的整理和回顾是必要的。
本文将涵盖课程内容的核心部分,帮助你更好地理解和记忆这些知识点。
需求与供给:理解需求曲线和供给曲线的概念及其影响因素,包括价格、收入、相关商品价格等。
掌握供需平衡时的市场价格及产量的决定。
弹性理论:理解弹性的概念及计算方法,掌握不同商品弹性的特性。
理解弹性与税收分配的关系。
消费者行为:理解边际效用理论和无差异曲线。
掌握消费者均衡的决定因素和消费者剩余的概念。
生产者行为:理解生产函数、成本函数和收益函数的概念。
掌握利润最大化的条件和边际收益与边际成本的关系。
国民收入核算:理解GDP的概念及核算方法,包括名义GDP和实际GDP。
掌握国民收入的基本公式及其实质含义。
通货膨胀与失业:理解通货膨胀的定义及衡量方法,掌握失业率的计算及类型。

天然药化知识点总结一、天然产物的来源天然产物是从自然界中获得的化合物,包括植物、微生物、动物等。
天然产物的来源非常广泛,可以是生长在地球上的任何一个角落。
1. 植物来源:植物是天然产物的重要来源之一。
许多植物都含有丰富的化学成分,如生物碱、多酚、鞣质、挥发油等,具有显著的药用价值。
例如,白芷、银杏、人参等都是常见的中药材。
此外,一些水果、蔬菜、谷物等食物中也含有许多具有药用价值的化合物。
2. 微生物来源:微生物也是天然产物的重要来源之一。
许多微生物具有生物合成能力,可以产生各种化合物,如抗生素、酶、氨基酸等。
青霉素、链霉素等就是从微生物中获得的抗生素。
3. 动物来源:动物也是天然产物的来源之一。
例如,海洋生物中含有丰富的活性成分,如海藻中的多醣体、海绵中的生物碱等。
此外,动物组织中也含有许多有益的化合物,如胰岛素、胰蛋白酶等。
二、天然产物的提取和分离技术天然产物的提取和分离是天然药化学研究的重要环节,它可以从天然产物中获得纯净的化合物,并为后续的结构鉴定、药效评价和药物改造提供原料。
1. 提取技术:提取是从天然产物中获得目标化合物的过程。
常用的提取方法有浸提、冷浸提、加热浸提、超临界流体萃取等。
其中,超临界流体萃取是一种新型的提取方法,它利用超临界流体的高溶解性和低表面张力,可以获得高纯度的提取物。
2. 分离技术:分离是从提取产物中获得纯净化合物的过程。
常用的分离技术有色谱法、透析法、结晶法、分液法等。
其中,色谱法是最常用的分离技术,包括薄层色谱、柱层析、高效液相色谱、气相色谱等。
三、天然药物的结构鉴定天然药物的结构鉴定是天然药化学研究的重要环节,它可以确定提取和分离得到的化合物的结构,为后续的药效评价和药物改造提供基础。
1. 光谱技术:光谱技术是天然药物结构鉴定的常用方法,包括紫外光谱、红外光谱、质谱、核磁共振等。
这些光谱技术可以从不同角度揭示化合物的结构信息,如它的功能团、分子式、分子量、分子结构等。
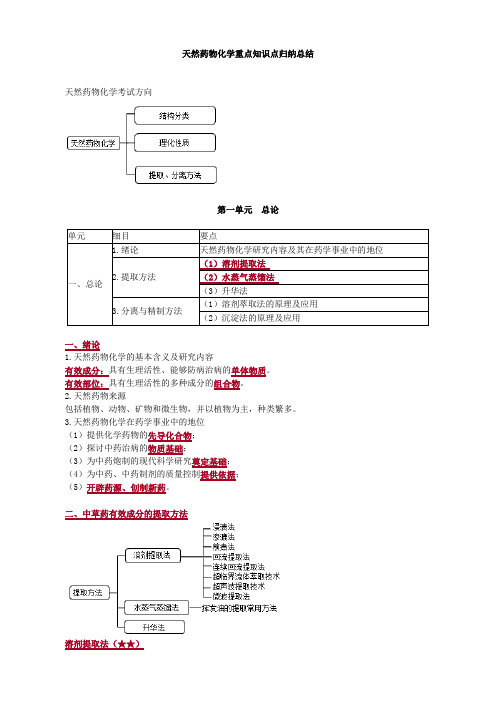
天然药物化学重点知识点归纳总结天然药物化学考试方向第一单元总论单元细目要点一、总论1.绪论天然药物化学研究内容及其在药学事业中的地位2.提取方法(1)溶剂提取法(2)水蒸气蒸馏法(3)升华法3.分离与精制方法(1)溶剂萃取法的原理及应用(2)沉淀法的原理及应用一、绪论1.天然药物化学的基本含义及研究内容有效成分:具有生理活性、能够防病治病的单体物质。
有效部位:具有生理活性的多种成分的组合物。
2.天然药物来源包括植物、动物、矿物和微生物,并以植物为主,种类繁多。
3.天然药物化学在药学事业中的地位(1)提供化学药物的先导化合物;(2)探讨中药治病的物质基础;(3)为中药炮制的现代科学研究奠定基础;(4)为中药、中药制剂的质量控制提供依据;(5)开辟药源、创制新药。
二、中草药有效成分的提取方法溶剂提取法(★★)1.溶剂选择1)常用的提取溶剂:亲脂性有机溶剂、亲水性有机溶剂和水。
常用中药成分提取的溶剂按极性由强到弱的顺序:水>甲醇>乙醇>丙酮>正丁醇>乙酸乙酯>二氯甲烷>乙醚>氯仿>苯>石油醚 巧记:水、甲乙丙丁蠢、只玩乙醚,仿苯室友 2)各类溶剂所能溶解的成分(相似相溶原理) 溶剂 类别可溶类型 具体类型水最安全,极性最强 能溶于水氨基酸、蛋白质、糖类、生物碱盐、有机酸盐、无机盐 甲醇(毒)、乙醇、丙酮亲水性有机溶剂 大极性的成分苷类、生物碱、鞣质及极性大的苷元正丁醇、乙酸乙酯、二氯甲烷、乙醚、氯仿、苯、石油醚 亲脂性有机溶剂 中等极性和小极性 生物碱、有机酸、蒽醌、黄酮、香豆素、强心苷 石油醚常用于脱脂,即通过溶解油脂、蜡、叶绿素小极性成分而将其与其他成分分开; 正丁醇是能与水分层的极性最大的有机溶剂,常用来从水溶液中萃取极性较大的苷类(皂苷)化合物。
溶剂提取方法 加热 提取溶剂 特点浸渍法 不水或其他提取时间长,效率不高渗漉法 不 水或醇 溶剂消耗量大,费时长煎煮法加 水含挥发性成分及加热易破坏的成分不宜使用回流提取法加有机溶剂 对热不稳定的成分不宜用此法,且消耗溶剂量大,操作麻烦连续回流提取法加有机溶剂 在实验室连续回流提取常采用索氏提取器或连续回流装置超临界流体萃取法:物质在临界温度和临界压力以上状态时常为单一相态,此单一相态称为超临界流体。

第一章1.主要的生物合成途径包含醋酸-丙二酸途径、甲戊二羟酸途径、桂皮酸途径及莽草酸途径、氨基酸途径和复合途径五种。
2.天然药物提取分离方法溶剂提取法、两相溶剂萃取法、沉淀法、盐析法、分馏法、结晶法、色谱法。
3.(了解)化合物的纯度测定4.(了解)结构研究的主要程序初步推断化合物类型→测定分子式,计算不饱和度→确定分子中含有的官能团,或结构片段,或基本骨架→推断并确定分子的平面结构→推断并确定分子的主体结构(构型、构象)5.(了解)结构测定常用的波谱分析紫外光谱,红外光谱,核磁共振谱(分为氢谱、碳谱、核磁共振新技术)、质谱、色谱-质谱连用技术第二章1.糖和苷的结构类型、性质及提取结构类型:单糖(monosaccharides) :多羟基醛和酮,不能再被简单地水解成更小分子的糖。
如葡萄糖、鼠李糖等。
低聚糖(oligosaccharides):单糖以半缩醛或半缩酮的形式以端基碳原子的羟基与另一分子糖结合而成。
由2~9个单糖聚合而成,也称为寡糖。
如蔗糖、麦芽糖等。
多糖(polysaccharides):类似于低聚糖。
由10个以上的单糖聚合而成,分子量很大。
其性质也大大不同于单糖和低聚糖。
如淀粉、纤维素等。
苷类:单糖以半缩醛或半缩酮的形式以端基碳原子的羟基与非糖物质缩合而成。
单糖一般为无色晶体,极易溶于水,多有甜味。
分子中有醛(酮)基、伯醇基、仲醇基和邻二醇基结构,易氧化。
如:银镜反应;硝基可使醛糖氧化成糖二酸;过碘酸氧化反应:主要作用于邻二醇羟基、α-氨基醇、α-羟基醛(酮)、α-羟基酸、邻二酮和某些活性次甲基结构。
具还原反应,成醛、成脂变旋光现象。
低聚糖性质与单糖近似,水溶性大,聚合度低的有甜味。
多糖无还原性,无变旋光现象,无甜味,大多难溶于水,有的能和水形成胶体溶液。
苷类多为固体,糖基少的可结晶,糖基多的则多为吸湿性的无定形粉末。
一般无味,但有的有苦味,很少的苷有甜味,溶解度随糖基数目增加而增加。
天然药物化学期末知识点整理天然药物化学是运用现代科学理论与方法研究天然药物中化学成分的一门学科。
以下是为大家整理的期末知识点,希望对大家的复习有所帮助。
一、天然药物化学成分的提取方法(一)溶剂提取法这是最常用的方法之一。
溶剂的选择至关重要,遵循“相似相溶”原则。
常用溶剂包括水、亲水性有机溶剂(如甲醇、乙醇)、亲脂性有机溶剂(如石油醚、氯仿)等。
浸渍法操作简便,但提取效率较低,适用于遇热不稳定成分的提取。
渗漉法提取效率高于浸渍法,但溶剂消耗量大。
煎煮法常用于提取水溶性成分,但对热不稳定成分不适用。
回流提取法和连续回流提取法效率较高,但需注意控制温度,避免成分破坏。
(二)水蒸气蒸馏法适用于具有挥发性、能随水蒸气蒸馏而不被破坏,且难溶或不溶于水的成分。
(三)升华法某些固体物质受热时直接气化,遇冷后又凝固为固体,此过程称为升华。
如樟木中的樟脑。
二、天然药物化学成分的分离方法(一)根据物质溶解度差异进行分离1、重结晶法:利用被提纯物质和杂质在溶剂中的溶解度差异,通过反复结晶来纯化。
2、沉淀法:通过加入试剂使有效成分或杂质生成沉淀而分离。
(二)根据物质在两相溶剂中的分配比不同进行分离1、液液萃取法:利用混合物中各成分在两种互不相溶的溶剂中分配系数的差异而达到分离。
2、纸色谱法:以滤纸为载体,以纸上所吸附的水为固定相,有机溶剂为流动相。
3、柱色谱法:包括硅胶柱色谱、氧化铝柱色谱、大孔吸附树脂柱色谱等。
(三)根据物质的吸附性差异进行分离1、物理吸附:如硅胶、氧化铝吸附。
硅胶适用于分离酸性和中性物质,氧化铝适用于分离碱性物质。
2、化学吸附:如聚酰胺吸附,对酚类、醌类、黄酮类等成分有较好的分离效果。
3、半化学吸附:如大孔吸附树脂,可用于分离水溶性成分。
(四)根据物质分子大小差异进行分离如凝胶过滤色谱法,常用的凝胶有葡聚糖凝胶、羟丙基葡聚糖凝胶等。
(五)根据物质解离程度不同进行分离离子交换色谱法,适用于分离具有解离性质的化合物。
天然药物化学重点总结1、主要生物合成途径醋酸——丙二酸(AA-MA):脂肪酸、酚类、蒽酮类脂肪酸:碳链奇数:丙酰辅酶A、支链:异丁酰辅酶A、α-甲基丁酰辅酶A、甲基丙二酸单酰辅酶A、碳链偶数:乙酰辅酶A甲戊二羟酸途径(MVA)桂皮酸途径和莽草酸途径氨基酸途径复合途径2、分配系数:两种相互不能任意混溶的溶剂K=C/ C(C 溶质在上相溶剂的浓度、C溶质在下相溶剂的浓度) UL UL3、分离难易度:A、B两种溶质在同一溶剂系统中分配系数的比值β=K/K (β>100一次萃取分离;10<β<100萃取10-12次;β<2一百以上;β=1不能分离) AB --+4、分配比与PHPH=pKa+lg[A]/[HA](pKa=[A][HO]/[HA]) 35、离子交换树脂阳离子交换树脂:交换出阳离子,交换碱性物质阴离子交换树脂:交换出阴离子,交换酸性物质1、几种糖的写法:D-木糖(Xyl)、D-葡萄糖(Glc)、D-甘露糖(Man)、D-半乳糖(Gal)、D-果糖(Flu)、L-鼠李糖(Rha)2、还原糖:具有游离醛基或酮基的糖非还原糖:不具有游离醛基或酮基的糖3、样品鉴别:样品+浓HSO+α-萘酚—?棕色环 244、羟基反应:醚化反应(甲醚化):Haworth法—可以全甲基话、Purdic法—不能用于还原糖、Kuhn法—可以部分甲基化、箱守法—可以全甲基化、反应在非水溶液中5、酸水解难易程度:N>O>S>C芳香属苷较脂肪属苷易水解:酚苷>萜苷、甾苷有氨基酸取代的糖较-OH糖难水解,-OH糖较去氧糖难水解(2,6二去氧糖>2-去氧糖>3-去氧糖>羟基糖>2-氨基糖)易?难呋喃糖苷较吡喃糖苷易水解酮糖较醛糖易水解吡喃糖苷中:C取代基越大越难水解(五碳糖>甲基五碳糖>六碳糖>七碳糖) 5 C上有-COOH取代时最难水解 5在构象中相同的糖中:a键(竖键)-OH多则易水解苷元为小基团—苷键横键比竖键易水解;即e>a苷元为大基团—苷键竖键比横键易水解;即a>e6、 smith降解(过碘酸反应):NaSO、NaBH,易得到苷元(人参皂苷—原人参二醇) 2447、乙酰解反应:β-苷键的葡萄糖双糖的反应速率(乙酰解反应的易难程度)(1——6)》(1——4)》(1——3)》(1——2)8、提取方法:水提醇沉、热水提冷水沉1、生物碱:生物碱是含负氧化态氮原子、存在于生物有机体的环状化合物。
天然药物化学是运用现代科学理论与方法研究天然药物中化学成分的一门学科1、溶剂提取法的基本原理:是根据 “相似者相溶”这一原理进行的,通过选择适当溶剂和方法将中药中的化学成分从药材中提取出来,溶剂法提取中药有效成分常用的方法,如浸渍法、渗漉法、煎煮法、回流提取法、连续回流提取法2、根据物质在两相溶剂中的分配比不同进行分离: 。
3、根据物质的吸附性差别进行分离4、根据物质分子大小差别进行分离:①分离天然化合物常用的方法有凝胶过滤法和膜分离技术;②常用的凝胶有葡聚糖凝胶 和羟丙基葡聚糖凝胶5、根据物质解离程度不同进行分离:2、糖的化学性质;氧化反应;糠醛形成反应(Molish 反应);羟基反应:醚化反应(甲基化)、酰化反应(酯化反应)、缩酮和缩醛化反应;羰基反应;和硼酸络合反应。
1、糖的提取分离②分离:活性炭柱色谱、 。
2、糖的鉴定和糖链结构的测定 。
1.苯丙素定义:一类含有一个或几个C6-C3单位的天然成分。
1.香豆素的基本母核:香豆素(香豆精)是具有苯并 -吡喃酮母核的一类化合物的总称。
环上常有-OH 、OCH 3、异戊烯基等取代基。
(1).简单香豆素类(2).呋喃香豆素类(furocoumarins)(线型和角型)(3).吡喃香豆素类(pyranocoumarins) (线型和角型)4.香豆素类化合物的提取分离 (1).提取O O 23456788a4a 回收溶剂加水苯EtOH O O H 3CO OCH 3O(2).分离主要包括:酸碱分离法,色谱方法等。
5.香豆素类化合物的结构鉴定(1).核磁法鉴定香豆素结构的意义:结构新颖的香豆素化合物不仅为创制新药提供了先导化合物,还为设计药效高、毒性低的理想药物提供了独特的化学结构,而核磁谱提供的信息是化合物结构鉴别的主要依据。
3.提取分离(1).提取:多用乙醇或丙酮等提取后,再用极性较小的溶剂如:乙醚、氯仿等进行萃取。
(2).分离:色谱法、溶剂萃取法、分级沉淀法、重结晶法.4.结构鉴定(1).化学法:水解反应、氧化反应。
天然药物化学总结绪论1、天然药物化学就是运用现代科学理论与方法研究天然药物中化学成分得一门学科。
研究内容:各类天然药物化学成分(主要就是生理活性成分或药效成分)得结构特点、物理化学性质、提取分离纯化方法、结构鉴定、生物合成途径。
2、天然药物:指人类在自然界中发现并可直接供药用得植物、动物、矿物、海洋生物、微生物等,以及基本不改变其药理化学属性得加工品。
3、(1)一次代谢产物(primary metabolites):糖类、脂质、蛋白质、核酸等对机体生命活动来说不可缺少得物质,普遍存在于动物、植物及微生物中。
(2)二次代谢产物(secondary metabolites):某个属、种或系统得生物所特有得,主要在植物、微生物中比较常见得物质。
这类化合物结构富于变化,多数具有明显得生理活性。
如生物碱、黄酮类、苷(甙)类、醌类、萜类、挥发油、苯丙素类、甾体类、鞣质、树脂、色素等。
4、天然药物得化学成分特点:(1)化学成分复杂;(2)具有多种临床用途。
分类:(1)有效成分(Active Constituents):经过不同程度得药效试验或生物活性试验,包括体外与体内试验,证明对机体具有一定生理活性得成分。
一般就是单体化合物:1、能用分子式与结构式表示;2、具有一定得理化常数;3、具有一定得生理活性。
(2)有效部位(Active Extracts):指具有生理活性得多种化学成分得混合物。
(3)无效成分:与有效成分共存得无生理活性得其它成分。
(4)有毒成分生物合成1、聚酮类化合物可根据分子结构中醋酸单位(C2单位)得数目进行命名,如聚庚酮类、聚己酮类等。
2、氨基酸途径作为前体得氨基酸:(1)脂肪族:鸟氨酸、赖氨酸(α-酮酸还原氨化生成)(2)芳香族:苯丙氨酸、酪氨酸、色氨酸(莽草酸途径生成)3、复合途径:(1)一个化合物分子有来自2个或2个以上不同生物合成途径得单元。
常见有:1、醋酸-丙二酸-莽草酸途径2、醋酸-丙二酸-甲戊二羟酸途径3、氨基酸-甲戊二羟酸途径4、氨基酸-醋酸-丙二酸途径5、氨基酸-莽草酸途径(2)一个化合物分子在不同植物中有不同得生物合成途径。
4、基本途径酮类途径过程产物单位甲戊二羟酸途径(MVA) 萜类、甾类、类胡萝卜素类C3桂皮酸途径苯丙氨酸经苯丙氨酸脱氨酶(PAL)脱氨后生成桂皮酸,进而得到苯丙素类化合物。
再经环化、氧化、还原等,可得到黄酮类(C6-C3-C6骨架)等化合物。
苯丙素类(C6-C3骨架)C6氨基酸途径(AA) 氨基酸脱羧成胺类,再经甲基化、氧化、还原、重排等系列反应得到生物碱。
本途径仅存在于植物、微生物中,就是其特有氮代谢方式。
生物碱AA提取分离方法1、准备工作(1)明确研究材料得学名、产地来源、采集时间与方法、使用部位(全体/部分)、材料得状态(新鲜品/干燥品)。
新鲜品不宜用与水不互溶得溶剂;而干燥品得干燥程度取决于目标化合物得稳定性。
一般情况下,样品要充分干燥、尽可能粉碎,提取效率高,提取操作、提取液得浓缩均较易进行。
注意样品不能粉碎过细,以20~60目为宜。
(2)明确研究目标提取分离已知成分或已知化学结构类型:查阅文献,找出该成分或该类结构类型成分得各种提取分离方案,再根据具体情况加以选用。
提取分离未知有效成分或有效部位:生理活性指导下提取分离;化学结构类型得理化性质指导下提取分离;根据化合物极性大小得不同系统提取分离。
2、注意问题(1)有效成分得丢失:含量高且有效、含量高且无效、含量低但有效(2)精制不纯:理化性质相近得化合物不易分离,对进一步得化学研究及药理试验产生影响。
3、提取(1)溶剂提取法1、根据天然药物中各种成分在不同溶剂中得溶解度不同,选用对有效成分溶解度大、对杂质成分溶解度小得溶剂,将有效成分从药材组织中溶解出来得方法。
溶剂通过扩散、渗透作用不断透过细胞壁、细胞膜进入细胞内,溶解可溶性成分,造成细胞内外得浓度差。
2、溶剂选择得依据——“相似相溶”常用提取溶剂及其极性强弱顺序:石油醚≈正己烷<四氯化碳<苯<二氯甲烷<无水乙醚<氯仿<乙酸乙酯<正丁醇<丙酮<乙醇<甲醇<水<吡啶<乙酸3、溶剂得选择原则根据材料得状态目标成分易溶,杂质成分难溶惰性,不与目标成分反应,或反应可逆经济安全、后续操作容易进行4、传统提取方法5、现代提取方法微波辅助提取法(microwave-assisted extraction)微波辅助提取就是利用微波能来提高提取率得一种新技术。
在微波场中,微波辐射导致植物细胞内得极性物质(水分子)吸收微波能,产生大量热量,使细胞内温度迅速上升,液态水汽化产生得压力将细胞膜与细胞壁冲破,形成微小得孔洞。
进一步加热,导致细胞内部与细胞壁水分减少,细胞收缩,表面出现裂纹。
孔洞与裂纹得存在使胞外溶剂容易进入细胞内,溶解并释放出胞内物质。
微波场中吸收微波能力得差异使得基体物质得某些区域或萃取体系中得某些组分被选择性加热,从而使得被萃取物质从基体或体系中分离,进入到介电常数较小、微波吸收能力相对较差得萃取剂中。
a)微波加热热效率高,升温快速而均匀,可显著缩短提取时间,提高提取效率。
b)设备简单、适用范围广、重现性好、节省试剂、污染小等。
超声提取法空化作用:当超声波在液体中传播时,使液体介质不断受到压缩与拉伸,拉伸时会在液体内部产生近似真空得小空洞;而压缩时,这些空洞发生崩溃,会使液体微粒间发生猛烈得撞击作用。
崩溃时空洞内部最高瞬时压力可达几万个大气压,同时还将产生局部得高温以及放电现象等。
微粒间这种剧烈得相互作用,起到很好得搅拌、分散,并使液体得温度骤然升高,从而使两种不相溶得液体发生乳化,加速溶质得溶解,促进化学反应。
利用超声波得空化作用,破坏细胞壁结构,使其在瞬间破裂,植物细胞内得成分得以释放,直接进入溶剂,加速植物有效成分得浸出提取;另外,超声波得次级效应,如机械振动、乳化、扩散、击碎、化学效应等也能加速欲提取成分得扩散释放并充分与溶剂混合,利于提取。
优点:提取时间短、提取效率高、无需加热等。
注意:提取瓶得放置位置、瓶壁得厚薄等会影响提取效果。
超临界流体萃取法超临界流体:温度、压力均高于临界点得流体。
其对物质得溶解能力随温度与压力得改变而在相当宽得范围内变动。
超临界流体相对接近液体得密度,使它有较高得溶解度;而其相对接近气体得粘度,使它具有较好得流动性、扩散性,对所需萃取得物质组织有较好得渗透性。
从而提高了溶质进入超临界流体得传质速率。
为提高选择性,可加入夹带剂。
优点:可以在近常温得条件下提取分离,几乎保留产品中全部有效成分,无有机溶剂残留,产品纯度高,操作简单、节能。
酶法提取通过酶反应较温与得将植物组织分解,加速有效成分得释放提取;将影响液体制剂得杂质如淀粉、蛋白质、果胶等分解去除,提高中药液体制剂得澄清度;将某些极性低得脂溶性成分转化为糖苷类易溶于水得成分,从而有利于提取。
注意:使用酶得浓度、底物、抑制剂、激动剂等;药材与水得比例、pH值、温度、时间等。
仿生提取法模拟口服药经胃肠道环境转运得原理而设计得提取方法。
尽可能地保留原药中得有效成分(包括在体内有效得代谢物、水解物、螯合物或新得化合物)。
6、系统溶剂分步提取根据提取要求、目得成分及杂质得性质差别、溶剂得溶解能力来确定提取方法:a)将固体药材按极性递增方式用不同溶剂依次提取。
石油醚、汽油:油脂、蜡、叶绿素、挥发油、游离甾体及三萜类等氯仿、乙酸乙酯:游离生物碱、有机酸、黄酮苷元、香豆素等丙酮、乙醇、甲醇:苷类、生物碱盐、鞣质等水:氨基酸、糖类、无机盐等水溶性成分b)将药材直接用乙醇、含水乙醇或含水丙酮提取,提取液浓缩成浸膏,拌以硅藻土等辅料,减压干燥成粉后,再用不同极性溶剂分步处理。
7、影响因素a)中草药成分间得相互作用对溶解度得影响(增溶、沉淀)b)粉碎度c)提取时间d)提取温度(2)水蒸气蒸馏法适用于具有挥发性得、能随水蒸汽蒸馏而不被破坏、与水不发生反应且难溶或不溶于水得成分得提取。
原理:两种互不相溶得液体共存时,其总蒸汽压等于各组分同一温度下蒸汽压之与。
由于体系得总蒸汽压比任一纯组分得蒸汽压高,所以混合物得沸点要比任一纯组分得沸点低。
(3)升华法适用于具有升华性质得化合物得提取与提纯。
简单易行,但回收不完全,并常伴有分解现象,产率低,适用于微量物质得精制,很少用于大规模生产制备。
(4)压榨法4、分离与精制(1)方法根据物质溶解度差别进行分离:结晶法、沉淀法、盐析法根据物质在两相溶剂中得分配比不同进行分离:液-液萃取法、逆流分溶法(CCD)、液滴逆流色谱法(DCCD)、高速逆流色谱法(HSCCC)、气液分配色谱法(GC或GLC)、液液分配色谱法(LC或LLC)根据物质得吸附性差别进行分离:硅胶、氧化铝、活性炭及聚酰胺、大孔吸附树脂根据物质分子大小差别进行分离:透析法、凝胶过滤法、超滤法、超速离心法根据物质解离程度不同进行分离:离子交换法、电泳(2)结晶法利用温度不同引起溶解度得改变以分离精制化合物得方法,就是分离精制固体化合物得重要方法之一。
通过重结晶进一步纯化化合物得结晶;纯化合物得结晶有一定得熔点与结晶学特征,有利于化合物性质及结构得判断;结晶法分离纯化得关键:结晶溶剂得选择与结晶条件。
1、结晶条件需结晶化合物在混合物中得含量选择合适得溶剂:单一/混合需结晶化合物在所选溶剂中得浓度合适得温度与时间需结晶化合物得性质:结晶性弱得化合物需要制备结晶性得衍生物或盐,如生物碱制为盐酸盐、氢溴酸盐、过氯酸盐、苦味酸盐等,羟基化合物制为乙酰化衍生物2、结晶溶剂选择理想得结晶溶剂需具备得条件:不与要结晶化合物起化学反应;选择性好;其它杂质在溶剂中得溶解度对温度得依赖性小,对杂质得溶解度非常大或非常小;溶剂得粘度要小,利于固液分离,且应使要结晶化合物容易成核,生成得晶体较完善。
如何选择:查阅文献,参考同类型化合物得一般溶解性质与结晶条件;根据要结晶化合物得极性大小,利用相似相溶规律通过实验选择溶剂;无理想单一溶剂时,可使用混合溶剂(一般选择对样品溶解度很大与很小得而又能够互溶得两种溶剂混合使用)。
3、结晶纯度得判断:晶形、色泽,熔点与熔距,薄层层析或纸层析,高效液相层析、气相层析。
(3)沉淀法提取物中加入某些试剂使产生沉淀,以获得有效成分或除去杂质得方法。
应注意得问题:沉淀得方法与技术要具有选择性;对于一些活性物质(如酶、蛋白质等)得沉淀分离,必须考虑沉淀方法对目标成分得活性与化学结构就是否破坏;残留物对人体得危害。
1、改变溶液中混合溶剂得极性水/醇法:沉淀除去糖类、蛋白质等水溶性杂质醇/水法:沉淀除去树脂、叶绿素等水不溶性杂质醇/醚法或醇/丙酮法:皂苷类成分沉淀析出2、改变溶液得pH酸/碱法:生物碱类成分得分离碱/酸法:黄酮、蒽醌类酚酸性成分得分离等电点沉淀法:蛋白质得分离3、加入沉淀试剂(如钙、钡、铅盐)(4)盐析法中草药得水提取液中,加入无机盐至一定浓度或达到饱与状态,使某些成分在水中得溶解度降低,沉淀析出或被有机溶剂提取出得分离方法。