价层电子对互斥理论
- 格式:doc
- 大小:189.50 KB
- 文档页数:5

第7讲价层电子对互斥理论根据预习里已经学习的内容,你发现常见分子或者离子的空间构型可以通过记忆获取。
但是,分子或者微粒的种类数目有许许多多,陌生微粒的空间构型应该如何推断呢?1.价层电子对互斥理论(VSEPR)分子中的价层电子对包括σ键电子对和中心原子上的孤电子对,由于电子对的相互排斥作用,而趋向尽可能彼此远离,分子尽可能采取对称的立体构型,以减小斥力。
2.价层电子对的确定方法(1)a 表示中心原子的价电子数。
对主族元素:a =最外层电子数; 对于阳离子:a =价电子数-离子电荷数; 对于阴离子:a =价电子数+|离子电荷数|。
(2)x 表示与中心原子结合的原子数。
(3)b 表示与中心原子结合的原子最多能接受的电子数,氢为1,其他原子=8-该原子的价电子数。
3.VSEPR 模型预测分子或离子的立体构型 (1)中心原子上的价电子都用于形成共价键的分子讲义一、导入二、知识讲解知识点1 价层电子对互斥理论平面三角形正四面体形(2)中心原子上有孤电子对的分子对于中心原子上有孤电子对(未用于形成共价键的电子对)的分子,中心原子上的孤电子对也要占据中心原子周围的空间,并互相排斥使分子呈现不同的立体构型。
VSEPR模型与分子的立体构型不一定一致,分子的立体构型指的是成键电子对的立体构型,不包括孤电子对(未用于形成共价键的电子对)。
两者是否一致取决于中心原子上有无孤电子对,当中心原子上无孤电子对时,两者的构型一致;当中心原子上有孤电子对时,两者的构型不一致。
三、例题精析【教学建议】此处内容主要用于教师课堂的精讲,每个题目结合试题本身、答案和解析部分,教师有的放矢的进行讲授或与学生互动练习。
例题11.用价层电子对互斥理论判断SO3的分子构型为()A.正四面体形B.V形C.三角锥形D.平面三角形解析:选D SO3中S原子的价层电子对数为3,其全部用于形成共价键,S原子周围有3个氧原子,属于平面三角形。
例题22.连线题。


价层电子对互斥理论使用价层电子对互斥理论是一种用来解释电子在能级上分布的理论。
该理论认为,在一个多电子系统中,电子倾向于占据不同的能级,这是因为电子之间存在一种互斥作用。
这种互斥作用源于电子之间的库仑排斥和泡利不相容原理。
库仑排斥是指电子之间由于带正电的原子核引力作用而产生的互斥力。
当两个电子靠近时,它们之间产生的排斥力会增加,使得电子倾向于占据不同的能级。
这种互斥作用对电子分布起到了显著的作用。
泡利不相容原理是指每个电子的量子态必须是唯一的。
根据泡利不相容原理,任意两个电子不能具有相同的量子态,即它们的自旋量子数必须不同。
这意味着电子不能全部集中在同一个能级上,而是倾向于分布在不同的能级上,以满足泡利不相容原理。
基于价层电子对互斥理论,可以解释许多电子结构的现象。
首先,可以解释原子的层次结构。
根据互斥理论,电子会填充不同的能级,从低能级开始,一直填充到高能级。
这解释了原子的能级结构和化学性质。
其次,互斥理论还可以解释原子和分子中电子的排布。
在原子中,电子遵循互斥原则,尽量占据不同的价层。
在分子中,电子在不同原子之间的共享和分配也遵循互斥原则。
这解释了化学键的形成和分子的几何结构。
互斥理论还有助于理解电子在固体中的行为。
固体中的电子被束缚在晶体中,组成电子云。
根据互斥理论,这些束缚电子也遵循互斥原则,尽量占据不同的能级。
这解释了导体、绝缘体和半导体的电子行为,以及电子在带隙里的分布。
此外,互斥理论还有助于解释多电子体系的能量分布。
根据互斥原理,电子将占据能量较低的态,使得多电子体系的总能量降低。
这解释了多电子体系的稳定性和能级分布。
总结来说,价层电子对互斥理论是一种解释电子在能级上分布的理论。
这一理论是基于库仑排斥和泡利不相容原理的,可以解释多种电子结构和化学现象。
它为我们理解原子、分子和固体的性质提供了重要的理论依据。

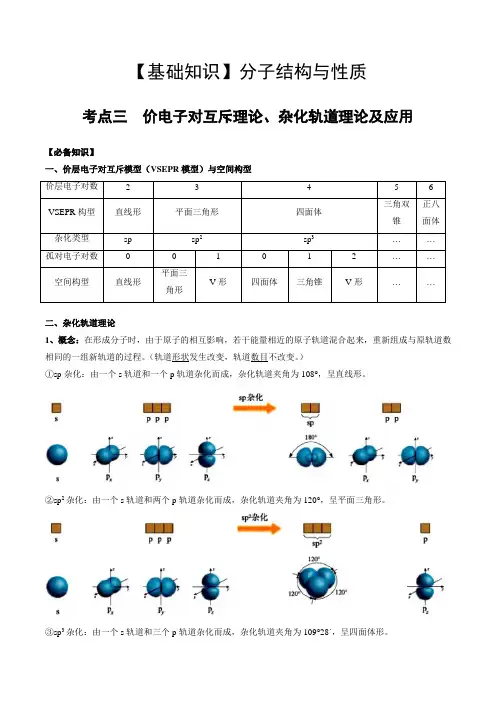
【基础知识】分子结构与性质考点三价电子对互斥理论、杂化轨道理论及应用【必备知识】一、价层电子对互斥模型(VSEPR模型)与空间构型价层电子对数 2 3 4 5 6VSEPR构型直线形平面三角形四面体三角双锥正八面体杂化类型sp sp2sp3……孤对电子对数0 0 1 0 1 2 ……空间构型直线形平面三角形V形四面体三角锥V形……二、杂化轨道理论1、概念:在形成分子时,由于原子的相互影响,若干能量相近的原子轨道混合起来,重新组成与原轨道数相同的一组新轨道的过程。
(轨道形状发生改变,轨道数目不改变。
)①sp杂化:由一个s轨道和一个p轨道杂化而成,杂化轨道夹角为108°,呈直线形。
②sp2杂化:由一个s轨道和两个p轨道杂化而成,杂化轨道夹角为120°,呈平面三角形。
③sp3杂化:由一个s轨道和三个p轨道杂化而成,杂化轨道夹角为109°28´,呈四面体形。
2、价电子对数=σ键电子对数+中心原子的孤电子对数说明:σ键电子对数=中心原子结合的原子数;中心原子的孤电子对数=1/2 (axb ±电荷)①a 表示中心原子的价电子数。
对于主族元素:a=原子的最外层电子数。
②x表示与中心原子结合的原子数。
③b表示与中心原子结合的原子最多能接受的电子数,即化合价绝对值。
例1、完成下列表格AB nб键数孤对电子对数价电子对数中心原子杂空间构型化轨道类型3 14 sp3三角锥形SOCl2中心原子为SNH4+ 4 0 4 sp3四面体SO42 4 0 4 sp3四面体ClO4 4 0 4 sp3四面体NH3 3 1 4 sp3三角锥形H2O 2 2 4 sp3V形SO3 3 0 3 sp2平面三角形BF3 3 0 3 sp2平面三角形SO2 2 1 3 sp2V形NO2 2 1 3 sp2V形CS2 2 0 2 sp 直线形O3 2 1 3 sp2平面三角形例2、判断以下有机物中原子的杂化轨道类型。





价层电子对互斥理论
双价层电子对互斥理论提出了一种新的观点,表明电子是双价的,而不是单价的,即双价层模型是由两个负责一个反应的活性立体复配,整个双价层电子互斥理论给出了一个反应前激子和过渡态之间的立体分子动力学框架,为理解有机分子反应的立体化学提供了基础性的理论支撑。
双价层电子对互斥理论和单价电子理论的核心正在于,电子活性的双价状态是有机反应的一个重要的特征,它们构成“立体复配”,也就是双价层模型,这种差异主要表现在双价层模型的跃迁态过程中的动力学行为上,即每一个轨道状态都只有一个立体结构,而且这两个轨道之间的不对称性也存在。
即使能量有所改变,这种不对称性也不会随之改变。
反应前激子和反应过渡态可以通过这种双轨道立体不对称性来解释,其中重要的是反应中的立体复配。

价层电子对互斥理论使用价层电子对互斥理论(Valence Shell Electron Pair Repulsion Theory)是一种用于推测分子几何形状的理论。
该理论假设电子对在空间中互相排斥,因此分子的几何构型可以通过电子对的排布来确定。
价层电子对是指与原子之间能够形成共价键或孤对电子的电子对。
理论基础:1.电子对之间的排斥力最小化。
同种电子对之间的排斥力较大,不同种电子对之间的排斥力较小。
2.单独的电子对比成对的电子对更容易被其他电子云所靠近。
3.电子对的排布趋向于最大程度的分离。
根据这些原则,我们可以预测分子的排布形态。
分子的电子对排布方式有多种形态,包括线性、平面三角形、平面四边形、平面五边形、笼形、六边形、八面体等。
以氨分子(NH3)为例,氮原子有5个电子,其中3个与3个氢原子形成共价键,剩下的2个是孤对电子。
根据价层电子对互斥理论,孤对电子会占据较大的空间,使得氨分子呈现出一个三角形构型。
再以甲烷(CH4)为例,碳原子有4个电子,与4个氢原子形成共价键。
根据价层电子对互斥理论,这4个电子对会互相排斥,使得甲烷分子呈现出一个正方形构型。
另外一个例子是水分子(H2O),氧原子有6个电子,与两个氢原子形成共价键,剩下的2个是孤对电子。
根据价层电子对互斥理论,这两个孤对电子会占据较大的空间,使得水分子呈现出一个倾斜的构型。
从上述示例可以看出,通过价层电子对互斥理论,我们可以推测出分子的几何构型,进而推断分子的性质和反应行为。
这为有机化学、无机化学以及生物化学等领域的研究提供了有力的理论基础。
然而,价层电子对互斥理论也有其局限性。
例如在一些分子中,价层电子对排布方式可能不符合预期,这可能是由于额外的电子云效应或分子内相互作用的影响。
此外,该理论只适用于描述小分子的几何构型,对于大分子或者有多个中心原子的分子来说并不适用。
尽管如此,价层电子对互斥理论依然是化学中一项重要的理论工具,为我们理解分子结构和性质提供了实质性的帮助,并促进了分子设计、药物研发以及催化反应等方面的进展。
价层电子对互斥理论价层电子对互斥理论(英文:V alence S hell E lectron P air R epulsion (VSEPR)),是一个用来预测单个共价分子形态的化学模型。
理论通过计算中心原子的价层电子数和配位数来预测分子的几何构型,并构建一个合理的路易斯结构式来表示分子中所有键和孤对电子的位置。
[编辑]理论基础价层电子对互斥理论的基础是,分子或离子的几何构型主要决定于与中心原子相关的电子对之间的排斥作用。
该电子对既可以是成键的,也可以是没有成键的(叫做孤对电子)。
只有中心原子的价层电子才能够对分子的形状产生有意义的影响。
分子中电子对间的排斥的三种情况为:孤对电子间的排斥(孤-孤排斥);孤对电子和成键电子对之间的排斥(孤-成排斥);成键电子对之间的排斥(成-成排斥)。
分子会尽力避免这些排斥来保持稳定。
当排斥不能避免时,整个分子倾向于形成排斥最弱的结构(与理想形状有最小差异的方式)。
孤对电子间的排斥被认为大于孤对电子和成键电子对之间的排斥,后者又大于成键电子对之间的排斥。
因此,分子更倾向于最弱的成-成排斥。
配体较多的分子中,电子对间甚至无法保持90°的夹角,因此它们的电子对更倾向于分布在多个平面上。
实际预测下面是价层电子对互斥理论预测的分子形状表。
没有孤电子对电子对数1个孤电子对2个孤电子对3个孤电子对(基本形状)2直线型3平面三角形型角型4四面体型三角锥型角型5三角双锥型变形四面体型T字型直线型6八面体型四角锥型平面四方形型7五角双锥型五角锥型8四方反棱柱型分子类型分子形状中心原子价电子对的排布方式†分子的几何构型‡实例AX1En双原子分子(直线型)HF、O2AX2E直线型BeCl2、HgCl2、CO2AX2E1角型NO2−、SO2、O3AX2E2角型H2O、OF2AX2E3直线型XeF2、I3−AX3E平面三角形型BF3、CO32−、NO3−、SO3AX3E1三角锥型NH3、PCl3AX3E2T字型ClF3、BrF3AX4E四面体型CH4、PO43−、SO42−、ClO4−AX4E1变形四面体型SF4AX4E2平面四方形型XeF4AX5E三角双锥型PCl5AX5E1四角锥型ClF5、BrF5AX6E八面体型SF6AX6E1五角锥型XeOF5−、IOF52−[1]AX7E五角双锥型IF7AX8E四方反棱柱型XeF2−8†孤电子对以淡黄色球体表示。
【高中化学】高中化学知识点:价层电子对互斥理论价层电子对互斥理论:1940年,美国sidgwicknv等人相继提出了价壳层电子对排斥理论(简称VSEPR方法),该理论适用于主族元素之间形成的abn类型分子或离子。
该理论认为,在共价分子或离子中,原子B(配位原子)围绕中心原子A的几何构型主要取决于中心原子价电子层中电子对之间的相互排斥。
这些电子对围绕中心原子尽可能远离彼此排列,以最小化彼此之间的排斥能。
所谓价电子对是指形成σ电子对和孤对键。
孤对电子的存在增加了电子对之间的斥力,影响了分子中的键角,并将改变分子构型的基本类型。
根据这一理论,只要已知分子或离子中中心原子上的价电子数,就可以轻松准确地判断ABn共价分子或离子的空间构型。
确定中心原子中价层电子对数:中心原子的价电子数与配体提供的公共电子数之和除以2即为中心原子的价电子对数。
规定:① 作为配体,卤素原子和氢原子提供一个电子,而氧元素的原子不提供电子;②作为中心原子,卤素原子按提供7个电子计算,氧族元素的原子按提供6个电子计算;③ 对于复合离子,在计算价电子的对数时,还应加上负离子的电荷数或减去正离子的电荷数;④计算电子对数时,若剩余1个电子,亦当作1对电子处理。
⑤ 双键、三键和其他多键被视为一对电子判断分子的空间构型:根据中心原子的价电子对数,从表1中找出相应的价电子对构型,然后根据价电子对中孤对电子的数量确定电子对的排列方式和分子的空间构型。
相关高中化学知识点:杂化轨道理论(中心原子杂化方式)混合轨道理论:是鲍林为了解释分子的立体结构提出的。
中心原子杂化轨道、孤电子对数及与之相连的原子数间的关系是:杂化轨道数=孤电子对数+与之相连的原子数。
杂化前后轨道总数比变,杂化轨道用来形成σ键或容纳孤对电子,未杂化的轨道与杂化轨道所在平面垂直,可用来形成π键。
常见的混合方法:(1)sp杂化:直线型如:co二、cs二(2)sp二杂化:平面三角形(等性杂化为平面正三角形)如:bcl三c二h四不等性杂化为v字型如:h二oh二sof二(3)sp三杂化:空间四面体(等性杂化为正四面体)如:ch 四、ccl四不等性杂化为三角锥如:nh三pcl三h三o+sp三d杂化:三角双锥服务提供商3d2混合体:八面体(与正八面体相等的混合体)分子的构型与杂化类型的关系:。
价层电子对互斥理论
价层电子对互斥理论(英文:V alence S hell E lectron P air R epulsion (VSEPR)),是一个用来预测单个共价分子形态的化学模型。
理论通过计算中心原子的价层电子数和配位数来预测分子的几何构型,并构建一个合理的路易斯结构式来表示分子中所有键和孤对电子的位置。
[编辑]理论基础
价层电子对互斥理论的基础是,分子或离子的几何构型主要决定于与中心原子相关的电子对之间的排斥作用。
该电子对既可以是成键的,也可以是没有成键的(叫做孤对电子)。
只有中心原子的价层电子才能够对分子的形状产生有意义的影响。
分子中电子对间的排斥的三种情况为:
∙孤对电子间的排斥(孤-孤排斥);
∙孤对电子和成键电子对之间的排斥(孤-成排斥);
∙成键电子对之间的排斥(成-成排斥)。
分子会尽力避免这些排斥来保持稳定。
当排斥不能避免时,整个分子倾向于形成排斥最弱的结构(与理想形状有最小差异的方式)。
孤对电子间的排斥被认为大于孤对电子和成键电子对之间的排斥,后者又大于成键电子对之间的排斥。
因此,分子更倾向于最弱的成-成排斥。
配体较多的分子中,电子对间甚至无法保持90°的夹角,因此它们的电子对更倾向于分布在多个平面上。
实际预测
下面是价层电子对互斥理论预测的分子形状表。
没有孤电子对(基本形状)1个孤电子对 2个孤电子对3个孤电子对
电子对数
2
直线型
3
平面三角形型角型
4
四面体型三角锥型角型
5
三角双锥型变形四面体型T字型直线型
6
八面体型四角锥型平面四方形型
7
五角双锥型五角锥型
8
四方反棱柱型
分子类
型分子形状
中心原子价电子对的排
布方式†
分子的几何
构型‡
实例
AX
1E
n
双原子分子
(直线型)
HF、O
2
AX
2E
直线型BeCl
2
、HgCl
2
、CO
2
AX
2E
1
角型NO
2
−、SO
2
、O
3
AX
2E
2
角型H
2
O、OF
2
AX
2E
3
直线型XeF
2
、I
3
−
AX
3E
平面三角形
型
BF
3
、CO
3
2−、NO
3
−、SO
3
AX
3E
1
三角锥型NH
3
、PCl
3
AX
3E
2
T字型ClF
3
、BrF
3
AX
4E
四面体型CH
4
、PO
4
3−、SO
4
2−、ClO
4
−
AX
4E
1
变形四面体
型
SF
4
AX
4E
2
平面四方形
型
XeF
4
AX
5E
三角双锥型PCl
5
AX
5E
1
四角锥型ClF
5
、BrF
5
AX
6E
八面体型SF
6
AX
6E
1
五角锥型XeOF
5
−、IOF
5
2−[1]
AX
7E
五角双锥型IF
7
AX
8E
四方反棱柱
型
XeF2−8
†孤电子对以淡黄色球体表示。
‡分子的实际几何构型,即不包含孤对电子的构型。
价层电子对互斥理论常用AXE方法计算分子构型。
这种方法也叫ABE,其中A代表中心原子,X或B代表配位原子,E代表孤电子对。
范例
甲烷分子(CH4)是四面体结构,是一个典型的AX4型分子。
中心碳原子周围有四个电子对,四个氢原子位于四面体的顶点,键角(H-C-H)为109°28'。
一个分子的形状不但受配位原子影响,也受孤对电子影响。
氨分子(NH3)中心原子杂化类型与甲烷相同(sp3),分子中有四个电子云密集区,电子云分布依然呈四面体。
其中三个是成键电子对,另外一个是孤对电子。
虽然它没有成键,但是它的排斥力影响着整个分子的形状。
因此,这是一个AX3E型分子,整个分子的形状是三角锥形,因为孤对电子是不可“见”的。
事实上,电子对数为七是有可能的,轨道形状是五角双锥。
但是它们仅存在于不常见的化合物之中,比如在六氟化氙中,有一对孤电子,它的构型趋向于八面体结构,因为孤对电子倾向于位于五角形的平面上。
另一个例子为七氟化碘,碘没有孤电子,七个氟原子呈五角双锥状排列。
电子对数为八也是有可能的,这些化合物一般为四方反棱柱体结构,[2]例子有八氟合氙酸亚硝酰中的 [XeF8]2−离子[3][4]以及八氰合钼(Ⅳ)阴离子 [Mo(CN)8]4−和八氟合锆(Ⅳ)阴离子 [ZrF8]4−。
例外
在一些化合物中VSEPR理论不能正确的预测分子空间构型。
过渡金属化合物
许多过渡金属化合物的几何构型不能用VSEPR理论解释,可以归结于价层电子中没有孤对电子以及核心的d电子与配体的相互作用。
[5]这些化合物的结构可以用VALBOND理论预测,包括金属氢化物和烷基配合物(例如六甲基钨),这个理论的基础是sd杂化轨道和三中心四电子键模型。
[6][7]晶体场理论是另一个经常可以解释配合物几何构型的理论。
IIA族卤化物
较重碱土金属的三原子卤化物的气相结构(例如:钙、锶、钡的卤化物,MX2)并不像预测的那样为直线型,而是V形。
(X-M-X的大致键角: CaF2,145°;SrF2,120°;BaF2,108°;SrCl2,130°;BaCl2,115°;BaBr2,115°;BaI2,105°).[8]格莱斯皮(Ronald Gillespie)提出这是因为配体与金属原子的内层电子发生相互作用,极化使得内层电子云不是完全球面对称,因此导致了分子构型的变化。
[5][9]
]一些AX2E2型分子
一个例子是氧化锂分子,即Li2O,它的中间构型是直线型而不是弯曲的,这一点可以归结于如果构型是弯曲的,锂原子之间将产生强烈的排斥作用。
[10]
另一个例子是O(SiH3)2(二甲硅醚)的Si-O-Si键的键角为144.1°,与其他分子中的键角相比差别较大,比如Cl2O (110.9°)、(CH3)2O (111.7°)以及N(CH3)3 (110.9°)。
格莱斯皮的合理解释是孤对电子的位置不同。
当配体的电负性与中心原子类似或更大时,孤对电子有能力排斥其他电子对,导致键角较小。
[5]当中心原子电负性较大时,就像O(SiH3)2中,孤对电子的定域不明显,排斥作用较弱,这种结合导致了强配体之间的排斥(-SiH3与上面的例子相比是一个比较大的配体)使得Si-O-Si键的键角比预想的要大。
[5]
一些形如AX6E1的分子
一些AX6E1型分子,例如含有Te(IV)或Bi(III)离子的化合物如TeCl62−、TeBr62−、BiCl63−、BiBr63−和BiI63−是正八面体结构;其孤对电子并不影响其构型[11]。
一种合理化解释是因为配体原子排列的拥挤没有给孤对电子留下空间[5];另一种合理化解释是惰性电子对效应。
[12]
与其他相关理论的对比
价层电子对互斥理论、价键理论和分子轨道理论都是关于分子如何构成的理论。
价键理论主要关注于σ键和π键的形成,通过研究受成键情况影响的轨道形状描述分子的形状。
价键理论也会借助VSEPR。
分子轨道理论则是关于原子和电子是如何组成分子或多原子离子的一个更精密的理论。