细菌内毒素USP
- 格式:docx
- 大小:49.84 KB
- 文档页数:12

美国药典24版细菌内毒素检查法本文是USP24细菌内毒素检查法(BACTERLAL ENDOTOXINS TEST)的译文,但目前执行的标准是USP24版第二增补本(USP—NF19 Second Supplement)的细菌内毒素检查法,读者可对照学习,从中可看出USP24细菌内毒素检查的不足之处。
本文将介绍用鲎试剂检测样品内或样品上存在的细菌内毒素浓度的方法。
鲎试剂(Limulus Amebocyte,LAL)来自鲎的循环细胞,经水提取而行,制备和检验后,用于凝固法。
反应的终点是将供检样品稀释后,与平行稀释的参考内毒素标准进行直接比较得到的。
测得的内毒素含量用规定的内毒素单位表示。
鲎试剂也是用浊度法或显色法来读取数据的,这些试齐只要能满足这些选择方法的要求,就可以使用。
在做试验时,需要先做出一条标准曲线,并用该曲线计算供检样品的内毒素含量,包括样品和对照液加鲎试剂后的培育时间和在分光光度计上读取光密度时使用的波长等操作,都应该事先规定好。
如果是终点浊度法,则到达培育期后,应立刻读取读数。
而动力学检测(凶手浊度法和显色法),光密度的测定是贯穿于整个反应期间,而用于计算结果的百分数就是从这些读数中计算出来的。
在用终点法的显色法检测时,在到达事先规定的反应时间后,添加停止酶反应的试剂使反应停止后,再读取数据。
1、参考内毒素标准与对照内毒素标准参考内毒素标准(Reference standard endotoxin,RSE)是美国药典(USP)的内毒素参考标准,其效价为每瓶10 000个USP内毒素单位(EU)。
每瓶参考内毒素标准加5ml鲎试剂用水(LAL Reagent Watet),用旋转搅拌器间歇搅拌30min使其溶解,制成原液,用这一原液制作合适的系列稀释。
原液应保存于冰箱中,用于以后的稀释,但保存时间不得超过14d。
用前,用旋转搅拌器强力搅拌至少3min,每一步稀释,至少搅拌30s,然后才能用它稀释下一步。

注射用水内毒素标准注射用水内毒素标准注射用水是指用于注射给人或动物使用的水。
它是一种非常重要的药物溶剂,对患者的生命安全有着至关重要的作用。
因此,注射用水的质量与安全性也备受关注。
其中,水中的毒素是评价注射用水质量的重要指标之一。
本文将介绍注射用水内毒素标准的相关内容。
一、注射用水内毒素的种类常见的注射用水内毒素包括细菌内毒素、霉菌内毒素、金属离子和有机物等。
其中,细菌内毒素和霉菌内毒素是注射用水中最主要的毒素种类。
二、细菌内毒素和霉菌内毒素1. 细菌内毒素细菌内毒素是一种有机物质,可导致严重的感染和炎症,危及患者的生命。
其中,内毒素A是最常见的细菌内毒素,可以引起休克、发热和多器官衰竭等严重并发症。
2. 霉菌内毒素霉菌内毒素也是一种有机物质,会导致各种毒性反应,如肝损伤、肾损伤和神经系统损伤等。
其中,黄曲霉素和赭曲霉素是较为常见的霉菌内毒素。
三、注射用水内毒素标准注射用水内毒素标准是由各国制订的规范,用于评价注射用水的毒素含量是否安全。
国际药典(PhEur)和美国药典(USP)是目前全球公认的注射用水内毒素标准。
1. 国际药典标准(PhEur)国际药典标准规定了注射用水中内毒素的含量限制。
其中,细菌内毒素的限制为0.5 EU/ml,而霉菌内毒素的限制为10 ng/ml。
2. 美国药典标准(USP)美国药典标准与国际药典标准相似,在注射用水中内毒素的含量限制上也有一定的规定。
其中,细菌内毒素的限制为0.25 EU/ml,而霉菌内毒素的限制为2 ng/ml。
四、注射用水内毒素检测方法注射用水内毒素检测方法是用于检测注射用水中毒素含量的方法,直接影响到注射用水的质量。
常用的检测方法包括生物法和化学方法。
1. 生物法生物法通过利用生物学检测原理,检测注射用水中内毒素的含量。
常见的生物法包括细菌内毒素检测和荧光素酶菌毒素检测。
2. 化学方法化学方法通过化学分析技术检测注射用水中内毒素的含量。
其中,高效液相色谱法和气相色谱法是目前比较常用的化学方法之一。



中国药典和美国药典的内毒素检测要求鲎试剂-LAL,中国药典和美国药典的内毒素检测要求鲎试剂是从栖⽣于海洋的节肢动物“鲎”的兰⾊⾎液中提取变形细胞溶解物,经低温冷冻⼲燥⽽成的⽣物试剂,专⽤于细菌内毒素检测。
鲎试验法是国际上⾄今为⽌检测内毒素最好的⽅法,它简单﹑快速﹑灵敏﹑准确,因⽽被欧美药典及我国药典定为法定内毒素检查法,并已被世界各国所采⽤。
⽬前国际上销售的鲎试剂有两种,⼀种称美洲鲎试剂(Limulus Amebocyte Lysate),缩写为LAL,由美国⽣产;另⼀种称东⽅鲎鲎试剂(Tachypleus Amebocyte Lysate),缩写为TAL。
实验证明:美洲鲎试剂-LAL⽐东⽅鲎试剂LAL更纯洁,检测效果更好。
TAL检查内毒素有很多⽅法,⽬前应⽤最⼴泛的是凝胶法,此外还有动态浊度法﹑显⾊基质法,⽐⾊法等。
其⽤途有以下⼏⽅⾯:1. 药检:⽤于注射药品﹑放射性药物﹑⽣物制品﹑注射器及⽣产⼯艺流程中的内毒素检测;2. 临床:⽤于检测病⼈各种体液中的内毒素含量;3. 其他:⽤于检测⽔或⾷品中的内毒素含量。
2005年版中国药典《细菌内毒素检查法-鲎试剂法》(鲎试剂-LAL法)本法系利⽤鲎试剂来检测或量化由⾰兰阴性菌产⽣的细菌内毒素,以判断供试品中细菌内毒素的限量是否符合规定的⼀种⽅法。
细菌内毒素检查包括两种⽅法,即凝胶法和光度测定法,后者包括浊度法和显⾊基质法。
供试品检测时,可使⽤其中任何⼀种⽅法进⾏试验。
当测定结果有争议时,除另有规定外,以凝胶法结果为准。
细菌内毒素的量⽤内毒素单位(EU)表⽰。
细菌内毒素国家标准品系⾃⼤肠杆菌提取精制⽽成,⽤于标定、复核、仲裁鲎试剂灵敏度和标定细菌内毒素⼯作标准品的效价。
细菌内毒素⼯作标准品系以细菌内毒素国家标准品为基准标定其效价,⽤于试验中鲎试剂灵敏度复核、⼲扰试验及设置的各种阳性对照。
凝胶法细菌内毒素检查⽤⽔系指内毒素含量⼩于0.015EU/ml的灭菌注射⽤⽔。

QCB细菌内毒素工作标准品标定操作规程QCB细菌内毒素工作标准品标定操作规程————————————————————————————————作者:————————————————————————————————日期:2标题:1.目的2.范围3.职责4.参考资料5.定义6.细菌内毒素工作标准品效价的标定标题:正文规范细菌内毒素工作标准品效价的标定程序,确保细菌内毒素检查结果的可靠性。
适用于质监处药理室对细菌内毒素工作标准品效价的标定。
质监处药理室分析人员负责本规程的实施和完成相关记录。
USP版附录85;CP版附录Ⅺ E 。
细菌内毒素检查法:本法系利用从鲎的变形细胞中提取的试剂来检测或量化由革兰阴性菌产生的细菌内毒素,以判断供试品中细菌内毒素的含量是否符合规定的一种方法。
细菌内毒素的量用内毒素单位(EU)表示。
鲎试剂:是鲎变形细胞的溶出物经提取而成。
鲎试剂灵敏度:在本检查法规定的条件下能检测出内毒素标准溶液或供试品溶液中的最低内毒素浓度,用EU/ml表示。
细菌内毒素检查用水(BET水):系指与灵敏度为0.03EU/ml 或更高灵敏度的鲎试剂在37±1 ?C 条件下24小时不产生凝集反应的灭菌注射用水。
细菌内毒素参考标准品(RSE):RSE 是USP细菌内毒素参考标准品,每瓶有10,000个USP 内毒素单位。
细菌内毒素工作标准品(CSE):是除RSE 之外的内毒素标准品。
反应终点浓度:是指系列递减的内毒素浓度中最后一个呈现阳性结果的浓度。
一、试剂、材料与仪器设备试剂:鲎试剂批号、灵敏度、规格、生产商。
细菌内毒素检查用水(BET水) 批号、规格、生产商。
细菌内毒素工作标准品(CSE) 批号、效价、生产商。
细菌内毒素参考标准品(RSE) 批号、效价、生产商。
材料及仪器设备:电热恒温水箱型号、设备编号。
电热鼓风干燥箱型号、设备编号。
正文漩涡混合器型号。
无热原试管规格:10?75mm。
无热原玻璃瓶规格:25ml。

BACTERIAL ENDOTOXINS TEST <85> USP35Portions of this general chapter have been harmonized with the corresponding texts ofthe European Pharmacopoeia and/or the Japanese Pharmacopoeia. Those portions that are not harmonized are marked with symbols () to specify this fact.这一章节已经和欧洲药典(EP)与日本药典(JP)统一。
没有统一的部分已经用标记出来。
The Bacterial Endotoxins Test (BET) is a test to detect or quantify endotoxins from Gram-negative bacteria using amoebocyte lysate from the horseshoe crab (Limulus polyphemus or Tachypleus tridentatus).细菌内毒素检查法(BET)是通过鲎的阿米巴细胞溶解产物来检测或定量来自革兰氏阴性菌的内毒素。
There are three techniques for this test: the gel-clot technique, which is based on gel formation; the turbidimetric technique, based on the development of turbidity after cleavage of an endogenous substrate; and the chromogenic technique, based on the development of color after cleavage of a synthetic peptide-chromogen complex. Proceed by any of the three techniques for the test. In the event of doubt or dispute, the final decision is made based upon the gel-clot technique unless otherwise indicated in the monograph for the product being tested. The test is carried out in a manner that avoids endotoxin contamination.细菌内毒素检查法有3种:凝胶法,基于凝胶的形成;浊度法,BACTERIAL ENDOTOXINS TEST USP32Portions of this general chapter have been harmonized with the corresponding texts of the European Pharmacopeia and/or the Japanese Pharmacopeia. Those portions that are notharmonized are marked with symbols () to specify this fact.This chapter provides a test to detect or quantify bacterial endotoxins that may be present in or on the sample of the article(s) to which the test is applied. It uses Limulus Amebocyte Lysate (LAL) obtained from the aqueous extracts of circulating amebocytes of horseshoe crab (Limulus polyphemus or Tachypleus tridentatus) which has been prepared and characterized for use as anLAL Reagent.1There are two types of techniques for this test: the gel-clot techniques, which are based on gel formation, and the photometric techniques. The latter include a turbidimetric method, which is based on the development of turbidity after cleavage of an endogenous substrate, and a chromogenic method, which is based on the development of color after cleavage of a synthetic peptide-chromogen complex. Proceed by any one of these techniques, unless otherwise indicated in the monograph. In case of dispute, the final decision is based on the gel-clot techniques, unless otherwise indicated in the monograph.In the gel-clot techniques, the reaction endpoint is determined from dilutions of the material under test in direct comparison with parallel dilutions of a reference endotoxin, and quantities of endotoxin are expressed in USP Endotoxin Units (USP-EU).[NOTE—One USP-EU is equal to one IU of endotoxin.]Because LAL Reagents have been formulated to be used also for turbidimetric or colorimetric tests, such tests may be used to comply with the requirements. These tests require the establishment of a standard regression curve; the endotoxin content of the test material is determined by interpolation from the curve. The procedures include incubation for a preselected time of reacting endotoxin and control solutions with LAL Reagent and reading of the spectrophotometric light absorbance at suitable wavelengths. In the endpoint turbidimetric procedure the reading is made immediately at the end of the incubation period. In the endpoint colorimetric procedure the reaction is arrested at the end of the preselected time by the addition of an enzyme reaction-terminating agent prior to the readings. In the turbidimetric and colorimetric kinetic assays the absorbance is measured throughout the reaction period and rate values are determined from those readings.APPARATUS AND GLASSWAREDepyrogenate all glassware and other heat-stable materials in a hot-air oven using a validatedprocess.2Commonly used minimum time and temperature settings are 30 minutes at 250. If employing plastic apparatus, such as microplates and pipet tips for automatic pipetters, useonly that which has been shown to be free of detectable endotoxin and not to interfere with the test.[NOTE—In this chapter, the term “tube” includes any other receptacle such as a micro-titer well.]PREPARATION OF THE STANDARD ENDOTOXIN STOCK SOLUTION AND STANDARD SOLUTIONS The USP Endotoxin RS has a defined potency of 10,000 USP Endotoxin Units (EU) per vial. Constitute the entire contents of 1 vial of the RSE with 5 mL of LAL Reagent Water3, mixintermittently for 30 minutes, using a vortex mixer, and use this concentrate for making appropriate serial dilutions. Preserve the concentrate in a refrigerator for making subsequent dilutions for not more than 14 days. Mix vigorously, using a vortex mixer, for not less than 3 minutes before use. Mix each dilution for not less than 30 seconds before proceeding to make the next dilution. Do not store dilutions, because of loss of activity by adsorption, in the absence of supporting data to the contrary.Preparatory TestingUse an LAL Reagent of confirmed label sensitivity.The validity of test results for bacterial endotoxins requires an adequate demonstration that specimens of the article or of solutions, washings, or extracts thereof to which the test is to be applied do not of themselves inhibit or enhance the reaction or otherwise interfere with the test. Validation is accomplished by performing the inhibition or enhancement test described under each of the three techniques indicated. Appropriate negative controls are included. Validation must be repeated if the LAL Reagent source or the method of manufacture or formulation of the article is changed.Preparation of Sample SolutionsPrepare sample solutions by dissolving or diluting drugs or extracting medical devices using LAL Reagent Water. Some substances or preparations may be more appropriately dissolved, diluted, or extracted in other aqueous solutions. If necessary, adjust the pH of the solution (or dilution thereof) to be examined so that the pH of the mixture of the LAL Reagent and sample falls within the pH range specified by the LAL Reagent manufacturer. This usually applies to a product with a pH in the range of 6.0 to 8.0. The pH may be adjusted using an acid, base, or suitable buffer as recommended by the LAL Reagent manufacturer. Acids and bases may be prepared from concentrates or solids with LAL Reagent Water in containers free of detectable endotoxin. Buffers must be validated to be free of detectable endotoxin and interfering factors.DETERMINATION OF MAXIMUM VALID DILUTION (MVD)The Maximum Valid Dilution is the maximum allowable dilution of a specimen at which the endotoxin limit can be determined. It applies to injections or to solutions for parenteral administration in the form constituted or diluted for administration, or, where applicable, to theamount of drug by weight if the volume of the dosage form for administration could be varied. The general equation to determine MVD is:MVD = (Endotoxin limit × Concentration of sample solution)/()where the concentration of sample solution and are as defined below. Where the endotoxin limit concentration is specified in the individual monograph in terms of volume (in EU per mL), divide the limit by, which is the labeled sensitivity (in EU per mL) of the LAL Reagent, to obtainthe MVD factor. Where the endotoxin limit concentration is specified in the individual monograph in terms of weight or Units of active drug (in EU per mg or in EU per Unit), multiply the limit by the concentration (in mg per mL or in Units per mL) of the drug in the solution tested or of the drug constituted according to the label instructions, whichever is applicable, and dividethe product of the multiplication by, to obtain the MVD factor. The MVD factor so obtained is the limit dilution factor for the preparation for the test to be valid.ESTABLISHMENT OF ENDOTOXIN LIMITSThe endotoxin limit for parenteral drugs, defined on the basis of dose, is equal to K/M,4where K is the threshold human pyrogenic dose of endotoxin per kg of body weight, and M isequal to the maximum recommended human dose of product per kg of body weight in a single hour period.The endotoxin limit for parenteral drugs is specified in individual monographs in units such as EU/mL, EU/mg, or EU/Unit of biological activity.GEL-CLOT TECHNIQUESThe gel-clot techniques detect or quantify endotoxins based on clotting of the LAL Reagent in the presence of endotoxin. The concentration of endotoxin required to cause the lysate to clot under standard conditions is the labeled sensitivity of the LAL Reagent. To ensure both the precision and validity of the test, tests for confirming the labeled LAL Reagent sensitivity and for interfering factors are described under Preparatory Testing for the Gel-Clot Techniques.Preparatory Testing for the Gel-Clot TechniquesTest for Confirmation of Labeled LAL Reagent Sensitivity—Confirm the labeled sensitivity using at least 1 vial of the LAL Reagent lot. Prepare a series of two-fold dilutions of the USP EndotoxinRS in LAL Reagent Water to give concentrations of 2,, 0.5, and 0.25, where is as defined above. Perform the test on the four standard concentrations in quadruplicate and。

BACTERIAL ENDOTOXINS TEST <85> USP35之马矢奏春创作Portions of this general chapter have been harmonized with the corresponding texts of the European Pharmacopoeia and/or the Japanese Pharmacopoeia. Those portions that are not harmonized are marked with symbols () to specify this fact.这一章节已经和欧洲药典(EP)与日本药典(JP)统一。
没有统一的部分已经用标识表记标帜出来。
The Bacterial Endotoxins Test (BET) is a test to detect or quantify endotoxins from Gram-negative bacteria using amoebocyte lysate from the horseshoe crab (Limulus polyphemus or Tachypleus tridentatus).细菌内毒素检查法(BET)是通过鲎的阿米巴细胞溶解产品来检测或定量来自革兰氏阴性菌的内毒素。
There are three techniques for this test: the gel-clot technique, which is based on gel formation; the turbidimetric technique, based on the development of turbidity after cleavage of an endogenous substrate; and the chromogenic technique, based on the development ofcolor after cleavage of a synthetic peptide-chromogen complex. Proceed by any of the three techniques for the test. In the event of doubt or dispute, the final decision is made based upon the gel-clot technique unless otherwise indicated in the monograph for the product being tested. The test is carried out in a manner that avoids endotoxin contamination.细菌内毒素检查法有3种:凝胶法,基于凝胶的形成;浊度法,BACTERIAL ENDOTOXINS TEST USP32Portions of this general chapter have been harmonized with the corresponding texts of the European Pharmacopeia and/or the Japanese Pharmacopeia. Those portions that are notharmonized are marked with symbols () to specify this fact.This chapter provides a test to detect or quantify bacterial endotoxins that may be present in or on the sample of the article(s) to which the test is applied. It uses Limulus Amebocyte Lysate (LAL) obtained from the aqueous extracts of circulating amebocytes of horseshoe crab (Limulus polyphemus or Tachypleus tridentatus) which has been prepared and characterized for use as anLAL Reagent.1There are two types of techniques for this test: the gel-clot techniques, which are based on gel formation, and the photometric techniques. The latter include a turbidimetric method, which is based on the development of turbidity after cleavage of an endogenous substrate, and a chromogenic method, which is based on the development of color after cleavage of a synthetic peptide-chromogen complex. Proceed by any one of these techniques, unless otherwise indicated in the monograph. In case of dispute, the final decision is based on the gel-clot techniques, unless otherwise indicated in the monograph.In the gel-clot techniques, the reaction endpoint is determined from dilutions of the material under test in direct comparison with parallel dilutions of a reference endotoxin, and quantities of endotoxin are expressed in USP Endotoxin Units (USP-EU). [NOTE—One USP-EU is equal to one IU of endotoxin.]Because LAL Reagents have been formulated to be used also for turbidimetric or colorimetric tests, such tests may be used to comply with the requirements. These tests require the establishment of a standard regression curve; the endotoxin content of the test material is determined by interpolation from the curve. The procedures include incubation for a preselected time of reacting endotoxin and control solutions with LAL Reagent and reading of the spectrophotometric light absorbance at suitable wavelengths. In the endpoint turbidimetric procedure the reading is made immediately at the end of the incubation period. In the endpoint colorimetric procedure the reaction is arrested at the end of the preselected time by the addition of an enzyme reaction-terminating agent prior to the readings. In the turbidimetric and colorimetric kinetic assays the absorbance is measured throughout the reaction period and rate values are determined from those readings.APPARATUS AND GLASSWAREDepyrogenate all glassware and other heat-stable materials in a hot-air oven using a validated process.2 Commonly used minimum time and temperature settings are 30 minutes at 250.If employing plastic apparatus, such as microplates and pipet tips for automatic pipetters, use only that which has been shown to be free of detectable endotoxin and not to interfere with the test. [NOTE—In th is chapter, the term “tube” includes any other receptacle such as a micro-titer well.]PREPARATION OF THE STANDARD ENDOTOXIN STOCK SOLUTION AND STANDARD SOLUTIONSThe USP Endotoxin RS has a defined potency of 10,000 USP Endotoxin Units (EU) per vial. Constitute the entire contents of 1 vial of the RSE with 5 mL of LAL Reagent Water3, mixintermittently for 30 minutes, using a vortex mixer, and use this concentrate for making appropriate serial dilutions. Preserve the concentrate in a refrigerator for making subsequent dilutions for not more than 14 days. Mix vigorously, using a vortex mixer, for not less than 3 minutes before use. Mix each dilution for not less than 30 seconds before proceeding to make the next dilution. Do not store dilutions, because of loss of activity by adsorption, in the absence of supporting data to the contrary.Preparatory TestingUse an LAL Reagent of confirmed label sensitivity.The validity of test results for bacterial endotoxins requires an adequate demonstration that specimens of the article or of solutions, washings, or extracts thereof to which the test is to be applied do not of themselves inhibit or enhance the reaction or otherwise interfere with the test. Validation is accomplished by performing the inhibition or enhancement test described under each of the three techniques indicated. Appropriate negative controls are included. Validation must be repeated if the LAL Reagent source or the method of manufacture or formulation of the article is changed.Preparation of Sample SolutionsPrepare sample solutions by dissolving or diluting drugs or extracting medical devices using LAL Reagent Water. Some substances or preparations may be more appropriately dissolved, diluted, or extracted in other aqueous solutions. If necessary, adjust the pH of the solution (or dilution thereof) to be examined so that the pH of the mixture of the LAL Reagent and sample falls within the pH range specified by the LAL Reagent manufacturer. This usually applies to a product with a pH in the range of 6.0 to 8.0. The pH may be adjusted using an acid, base, or suitable buffer as recommended by the LAL Reagent manufacturer. Acids and bases may be prepared from concentrates or solids with LAL Reagent Water in containers free of detectable endotoxin. Buffers must be validated to be free of detectable endotoxin and interfering factors.DETERMINATION OF MAXIMUM VALID DILUTION (MVD)The Maximum Valid Dilution is the maximum allowable dilution of a specimen at which the endotoxin limit can be determined. It applies to injections or to solutions for parenteral administration in the form constituted or diluted for administration, or, where applicable, to the amount of drug by weight if the volume of the dosage form for administration could be varied. The general equation to determine MVD is:MVD = (Endotoxin limit × Concentration of sample solution)/()where the concentration of sample solution and are as defined below. Where the endotoxin limit concentration is specified in the individual monograph in terms of volume (in EU per mL),divide the limit by , which is the labeled sensitivity (in EU per mL) of the LAL Reagent, to obtainthe MVD factor. Where the endotoxin limit concentration is specified in the individual monograph in terms of weight or Units of active drug (in EU per mg or in EU per Unit), multiply the limit by the concentration (in mg per mL or in Units per mL) of the drug in the solution tested or of the drug constituted according to the label instructions, whichever is applicable, and divide theproduct of the multiplication by , to obtain the MVD factor. The MVD factor so obtained is thelimit dilution factor for the preparation for the test to be valid.ESTABLISHMENT OF ENDOTOXIN LIMITSThe endotoxin limit for parenteral drugs, defined on the basis of dose, is equal to K/M,4 where K is the threshold human pyrogenic dose of endotoxin per kg of body weight, and M isequal to the maximum recommended human dose of product per kg of body weight in a single hour period.The endotoxin limit for parenteral drugs is specified in individual monographs in units such as EU/mL, EU/mg, or EU/Unit of biological activity.GEL-CLOT TECHNIQUESThe gel-clot techniques detect or quantify endotoxins based on clotting of the LAL Reagent in the presence of endotoxin. The concentration of endotoxin required to cause the lysate to clot under standard conditions is the labeled sensitivity of the LAL Reagent. To ensure both the precision and validity of the test, tests for confirming the labeled LAL Reagent sensitivity and for interfering factors are described under Preparatory Testing for the Gel-Clot Techniques.Preparatory Testing for the Gel-Clot TechniquesTest for Confirmation of Labeled LAL Reagent Sensitivity— Confirm the labeled sensitivity using at least 1 vial of the LAL Reagent lot. Prepare a series of two-fold dilutions of the USP EndotoxinRS in LAL Reagent Water to give concentrations of 2, , where is as defined above.Perform the test on the four standard concentrations in quadruplicate and include negative controls. The test for confirmation of lysate sensitivity is to be carried out when a new batch of LAL Reagent is used or when there is any change in the experimental conditions that may affect the outcome of the test.Mix a volume of the LAL Reagent with an equal volume (such as 0.1-mL aliquots) of one of the standard solutions in each test tube. When single test vials or ampuls containing lyophilized LAL Reagent are used, add solutions directly to the vial or ampul. Incubate the reaction mixture for aconstant period according to directions of the LAL Reagent manufacturer (usually at 37 ± 1 for 60 ± 2 minutes), avoiding vibration. To test the integrity of the gel, take each tube in turn directly from the incubator and invert it through about 180 in one smooth motion. If a firm gel hasformed that remains in place upon inversion, record the result as positive. A result is negative if an intact gel is not formed. The test is not valid unless the lowest concentration of the standard solutions shows a negative result in all replicate tests.The endpoint is the last positive test in the series of decreasing concentrations of endotoxin. Calculate the mean value of the logarithms of the endpoint concentration and then the antilogarithm of the mean value using the following equation:Geometric Mean Endpoint Concentration = antilog (Se / f)and not more than 2, the labeled sensitivity is confirmed and is used in tests performed withthis lysate.Interfering Factors Test for the Gel-Clot Techniques— Prepare solutions A, B, C, and D as shown in Table 1, and perform the inhibition/enhancement test on the sample solutions at a dilution less than the MVD, not containing any detectable endotoxins, following the procedure in the Test for Confirmation of Labeled LAL Reagent Sensitivity above. The geometric mean endpoint concentrations of solutions B and C are determined using the equation in that test.Table 1. Preparation of Solutions for the Inhibition/Enhancement Test for Gel-Clot TechniquesSolutionEndotoxinConcentration/Solution towhich Endotoxin is Added DiluentDilutionFactorInitialEndotoxin ConcentrationNumberofReplicatesAa none/sample solution ——— 4Bb 2/sample solution samplesolution1 2 42 1 44 48 4Cc 2/water for BET LALReagentWater1 2 22 1 24 28 2SolutionEndotoxinConcentration/Solution towhich Endotoxin is Added DiluentDilutionFactorInitialEndotoxin ConcentrationNumberofReplicatesDd none/LAL Reagent Water ——— 2a Solution A: a sample solution of the preparation under test that is free of detectable endotoxins.b Solution B: test for interference.c Solution C: control for labeled LAL Reagent sensitivity.d Solution D: negative control of LAL Reagent Water.This test must be repeated when any condition that is likely to influence the test results changes. The test is not valid unless Solutions A and D show no reaction and the result of Solution C confirms the labeled sensitivity.and not greater than 2, the sample solution does not contain factors which interfere underthe experimental conditions used. Otherwise, the sample solution to be examined interferes with the test.If the sample under test does not comply with the test at a dilution less than the MVD, repeat the test using a greater dilution, not exceeding the MVD. The use of a more sensitive lysate permits a greater dilution of the sample to be examined and this may contribute to the elimination of interference.Interference may be overcome by suitable treatment, such as filtration, neutralization, dialysis, or heating. To establish that the chosen treatment effectively eliminates interference without loss of endotoxins, perform the assay described below using the preparation to be examined towhich USP Endotoxin RS has been added and which has been subjected to the selected treatment.Gel-Clot Limit TestThis test is used when a monograph contains a requirement for endotoxin limits.Procedure— Prepare Solutions A, B, C, and D as shown in Table 2, and perform the test on these solutions following the procedure in the Test for Confirmation of Labeled LAL Reagent Sensitivity under Preparatory Testing for the Gel-Clot Techniques.Table 2. Preparation of Solutions for the Gel-Clot Limit TestSolution* Endotoxin Concentration/Solution to which Endotoxin is AddedNumber ofReplicatesSolution* Endotoxin Concentration/Solution to which Endotoxin is AddedNumber ofReplicatesA none/diluted sample solution 2B 2/diluted sample solution 2C 2/LAL Reagent Water 2D none/LAL Reagent Water 2* Prepare Solution A and positive product control Solution B using a dilution not greater than the MVD and treatments as directed in the Interfering Factors Test for the Gel-Clot Techniques under Preparatory Testing for the Gel-Clot Techniques. Positive control Solutions B and C contain the standard endotoxin preparation at a concentration corresponding to twice the labeled LAL Reagent sensitivity. The negative control Solution D is LAL Reagent Water.Interpretation— The test is not valid unless both replicates of positive control Solutions B and C are positive and those of negative control Solution D are negative. The preparation under test complies with the test when a negative result is found for both tubes containing Solution A. The preparation under test does not comply with the test when a positive result is found for both tubes containing Solution A.Repeat the test when a positive result is found for 1 tube containing Solution A and a negative result for the other one. The preparation under test complies with the test when a negative result is found for both tubes containing Solution A in the repeat result. If the test is positive for the preparation under test at a dilution less than the MVD, the test may be repeated at a dilution not greater than the MVD.Gel-Clot AssayThis assay quantifies bacterial endotoxins in sample solutions by titration to an endpoint.Procedure— Prepare Solutions A, B, C, and D as shown in Table 3, and test these solutions by following the procedure in the Test for Confirmation of Labeled LAL ReagentSensitivity under Preparatory Testing for the Gel-Clot Techniques.Table 3. Preparation of Solutions for the Gel-Clot AssaySolutionEndotoxinConcentration/Solution towhich Endotoxin is Added DiluentDilutionFactorInitialEndotoxinConcentrationNumberofReplicatesAa none/sample solution LALReagentWater1 — 2SolutionEndotoxinConcentration/Solution towhich Endotoxin is Added DiluentDilutionFactorInitialEndotoxinConcentrationNumberofReplicates2 — 24 — 28 — 2Bb 2/sample solution — 1 2 2Cc 2/LAL Reagent Water LALReagentWater1 2 22 1 24 28 2Dd none/LAL Reagent Water ——— 2a Solution A: a sample solution under test at the dilution, not to exceed the MVD, with which the Interfering Factors Test for the Gel-Clot Techniques was completed. Subsequent dilution of the sample solution must not exceed the MVD. Use LAL Reagent Water to make dilution series of four tubes containing the sample solution under test at concentrations of 1, ½, ¼, and 1/8 relative to the dilution with which the Interfering Factors Test for the Gel-Clot Techniques was completed. Other dilutions may be used as appropriate.b Solution B: Solution A containing standard endotoxin at a concentration of 2 (positive product control).c Solution C: two series of 4 tubes of LAL Reagent Water containing the standard endotoxin at a concentration of 2, , respectively.d Solution D: LAL Reagent Water (negative control).to 2.To determine the endotoxin concentration of Solution A, calculate the endpoint concentration for each replicate series of dilutions by multiplying each endpoint dilution factor by . Theendotoxin concentration in the sample is the geometric mean endpoint concentration of the replicates (see the formula given in the Test for Confirmation of Labeled LAL Reagent Sensitivity under Preparatory Testing for the Gel-Clot Techniques). If the test is conducted with a diluted sample solution, calculate the concentration of endotoxin in the original sample solution by multiplying by the dilution factor. If none of the dilutions of the sample solution is positive in avalid assay, report the endotoxin concentration as less than (if the diluted sample was tested,less than times the lowest dilution factor of the sample.) If all dilutions are positive, the endotoxin concentration is reported as equal to or greater than the greatest dilution factor multiplied by (e.g., initial dilution factor times 8 times in Table 3).The article meets the requirements of the test if the concentration of endotoxin is less than that specified in the individual monograph.PHOTOMETRIC TECHNIQUESThe turbidimetric method measures increases in turbidity. Depending on the test principle used, this technique is classified as either endpoint-turbidimetric or kinetic-turbidimetric. The endpoint-turbidimetric technique is based on the quantitative relationship between the concentration of endotoxins and the turbidity (absorbance or transmission) of the reaction mixture at the end of an incubation period. The kinetic-turbidimetric technique is a method to measure either the onset time needed to reach a predetermined absorbance of the reaction mixture or the rate of turbidity development.The chromogenic method measures the chromophore released from a suitable chromogenic peptide by the reaction of endotoxins with the LAL Reagent. Depending on the test principle employed, this technique is classified as either endpoint-chromogenic or kinetic-chromogenic. The endpoint-chromogenic technique is based on the quantitative relationship between the concentration of endotoxins and the release of chromophore at the end of an incubation period. The kinetic-chromogenic technique is a method to measure either the onset time needed to reach a predetermined absorbance of the reaction mixture or the rate of color development.All photometric tests are carried out at the incubation temperature recommended by the LAL Reagent manufacturer, which is usually 37 ± 1.Preparatory Testing for the Photometric TechniquesTo assure the precision or validity of the turbidimetric and chromogenic techniques, preparatory tests are conducted to verify that the criteria for the standard curve are valid and that the sample solution does not inhibit or enhance the reaction. Revalidation for the test method is required when conditions that are likely to influence the test result change.Verification of Criteria for the Standard Curve— Using the Standard Endotoxin Solution, prepare at least three endotoxin concentrations to generate the standard curve. Perform the test using at least three replicates of each standard endotoxin concentration according to the manufacturer's instructions for the LAL Reagent (with regard to volume ratios, incubation time, temperature, pH,etc.). If the desired range in the kinetic methods is greater than two logs, additional standards should be included to bracket each log increase within the range of the standard curve. The absolute value of the correlation coefficient, |r|, must be greater than or equal to 0.980 for the range of endotoxin concentrations indicated by the manufacturer of the LAL Reagent.Interfering Factors Test for the Photometric Techniques— Select an endotoxin concentration at or near the middle of the endotoxin standard curve. Prepare Solutions A, B, C, and D as shownin Table 4. Perform the test on Solutions A, B, C, and D at least in duplicate following the instructions for the LAL Reagent used (with regard to volume of sample and LAL Reagent, volume ratio of sample to LAL Reagent, incubation time, etc.).Table 4. Preparation of Solutions for the Inhibition/Enhancement Test for Photometric TechniquesSolution Endotoxin ConcentrationSolution to whichEndotoxin is AddedNumber ofReplicatesAa none sample solution not less than 2Bb middle concentration of the standardcurvesample solution not less than 2Cc at least 3 concentrations (lowest concentration is designated) LAL Reagent Water each not lessthan 2Dd none LAL Reagent Water not less than 2a Solution A: the sample solution may be diluted not to exceed MVD.b Solution B: the preparation under test at the same dilution as Solution A, containing added endotoxin at a concentration equal to or near the middle of the standard curve.c Solution C: the standard endotoxin at the concentrations used in the validation of the method described in Verification of Criteria for the Standard Curve under Preparatory Testing for the Photometric Techniques (positive control series).d Solution D: LAL Reagent Water (negative control).Calculate the mean recovery of the added endotoxin by subtracting the mean endotoxin concentration in the solution (if any) from that containing the added endotoxin. In order to be considered free of interfering factors under the conditions of the test, the measured concentration of the endotoxin added to the sample solution must be within 50% to 200% of the known added endotoxin concentration after subtraction of any endotoxin detected in the solution without added endotoxin.When the endotoxin recovery is out of the specified ranges, the interfering factors must be removed as described in the Interfering Factors Test for the Gel-ClotTechniques under Preparatory Testing for the Gel-Clot Techniques. Repeating the Interfering Factors Test for the Gel-Clot Techniques validates the treatment.Procedure for the Photometric TechniquesFollow the procedure described in the Interfering Factors Test for the Photometric Techniques under Preparatory Testing for the Photometric Techniques.Calculation for the Photometric TechniquesCalculate the endotoxin concentration of each of the replicates of test Solution A using the standard curve generated by positive control series C. The test is not valid unless the following conditions are met: (1) the results of control series C comply with the requirements for validation defined under Verification of Criteria for the Standard Curve under Preparatory Testing for the Photometric Techniques; (2) the endotoxin recovery, calculated from the concentration found in Solution B after subtracting the endotoxin concentration found in Solution A is within 50 to 200%; and (3) the result of negative control series D does not exceed the limit of the blank value required in the description of the LAL Reagent used.Interpretation of Results from the Photometric TechniquesIn photometric assays, the preparation under test complies with the test if the mean endotoxin concentration of the replicates of Solution A, after correction for dilution and concentration, is less than the endotoxin limit for the product.1 LAL Reagent reacts with some -glucans in addition to endotoxins. Some preparations that are treated will not react with -glucans and must be used for samples that contain glucans.2 For a validity test of the procedure for inactivating endotoxins, see Dry-HeatSterilization under Sterilization and Sterility Assurance of Compendial Articles 1211. Use an LAL Reagent having a sensitivity of not less than 0.15 Endotoxin Unit per mL.3 Sterile Water for Injection or other water that shows no reaction with the specific LAL Reagent with which it is to be used, at the limit of sensitivity of such reagent.4 K is5 USP-EU/kg for any route of administration other than intrathecal (for which K is 0.2USP-EU/kg body weight). For radiopharmaceutical products not administered intrathecally the endotoxin limit is calculated as 175/V, where V is the maximum recommended dose in mL. For intrathecally administered radiopharmaceuticals, the endotoxin limit is obtained by the formula14/V. For formulations (usually anticancer products) administered on a per square meter of body surface, the formula is K/M, where K = 5 EU/kg and M is the (maximum dose/m2/hour × 1.80m2)/70 Kg.。

再论细菌内毒素限值的确定中国药品生物制品检定所北京 100050王思理张国来蔡彤国家药典委员会北京 100061王平张莜红中国药典2000年版已经正式出版,并将于2000年7月1日正式执行。
细菌内毒素检查法应用指导原则及附件细菌内毒素定量检测法(动态浊度法和显色基质法)均被列入新版药典。
指导原则中对细菌内毒素限值的确定有非常明确清楚的规定:"细菌内毒素限值的确定药品、生物制品的细菌内毒素限值(L)一般按以下公式确定:L=K/M式中 L为供试品的细菌内毒素限值,以EU/ml、EU/mg、EU/u表示。
K为按规定的给药途径,人用每公斤体重每小时最大可接受的内毒素剂量,以EU/(kg·hr)表示。
注射剂,K=5EU/(kg·hr);其中放射性药品注射剂,K=2.5 EU/(kg·hr);鞘内用注射剂,K=0.02 EU/(kg·hr)。
M为人用每公斤体重每小时最大剂量,以ml/ (kg·hr)、mg/(kg·hr)、u/(kg·hr)表示,人均体重按60kg计算,注射时间小于1小时,按1小时计算。
按人用剂量计算L值时,如遇特殊情况,可根据生产和临床用药实际作必要调整。
应该说,今后国内对细菌内毒素限值(L)的确定可以统一了。
然而,从目前发表的文章和笔者审核的成果报告等都发现在药品的细菌内毒素检查法建立时对关键性的细菌内毒素限值(L)的确定尚存在不少问题。
主要问题是对"M为人用每公斤体重每小时最大剂量"作了如下的解释:其一、把最大剂量解释成"热原检查法剂量"。
例如:"注射用硝普钠中细菌内毒素的检查注射用硝普钠的内毒素限值(L):根据药品内毒素限值的计算方法,K=5.0EU/Kg,注射用硝普钠热原检查剂量M=1mg/KgL=K/M=(5.0EU/Kg)/(1mg/Kg)=5EU/mg。

再论细菌内毒素限值的确定中国药品生物制品检定所北京 100050王思理张国来蔡彤国家药典委员会北京 100061王平张莜红中国药典2000年版已经正式出版,并将于2000年7月1日正式执行。
细菌内毒素检查法应用指导原则及附件细菌内毒素定量检测法(动态浊度法和显色基质法)均被列入新版药典。
指导原则中对细菌内毒素限值的确定有非常明确清楚的规定:"细菌内毒素限值的确定药品、生物制品的细菌内毒素限值(L)一般按以下公式确定:L=K/M式中 L为供试品的细菌内毒素限值,以EU/ml、EU/mg、EU/u表示。
K为按规定的给药途径,人用每公斤体重每小时最大可接受的内毒素剂量,以EU/(kg·hr)表示。
注射剂,K=5EU/(kg·hr);其中放射性药品注射剂,K=2.5 EU/(kg·hr);鞘内用注射剂,K=0.02 EU/(kg·hr)。
M为人用每公斤体重每小时最大剂量,以ml/ (kg·hr)、mg/(kg·hr)、u/(kg·hr)表示,人均体重按60kg计算,注射时间小于1小时,按1小时计算。
按人用剂量计算L值时,如遇特殊情况,可根据生产和临床用药实际作必要调整。
应该说,今后国内对细菌内毒素限值(L)的确定可以统一了。
然而,从目前发表的文章和笔者审核的成果报告等都发现在药品的细菌内毒素检查法建立时对关键性的细菌内毒素限值(L)的确定尚存在不少问题。
主要问题是对"M为人用每公斤体重每小时最大剂量"作了如下的解释:其一、把最大剂量解释成"热原检查法剂量"。
例如:"注射用硝普钠中细菌内毒素的检查注射用硝普钠的内毒素限值(L):根据药品内毒素限值的计算方法,K=5.0EU/Kg,注射用硝普钠热原检查剂量M=1mg/KgL=K/M=(5.0EU/Kg)/(1mg/Kg)=5EU/mg。
生物检测实验<85> 细菌内毒素检测♦本文中部分内容与欧洲药典和/或日本药典的相应的内容是一致的,那些不一致的内容则用符号(♦♦)标明。
♦本文提供一种检测或定量可能出现在样品中的细菌内毒素的实验,它使用从鲎(美洲鲎或东方鲎)的循环阿米巴细胞的抽提液中制备的鲎试剂。
♦1细菌内毒素实验有两种技术:基于凝胶形成的凝胶法和光度测定法。
后一种方法包括浊度法——基于一内源性基质断裂后的浊度变化;和显色基质法——基于一个带有生色团的合成肽断裂后颜色的变化。
除非文中另有说明,否则可用上述方法中的任何一种。
若检测结果存在争议,除非文中另有说明,以凝胶法结果为准。
在凝胶法技术中,反应的终点是将供试品稀释液与平行稀释的参考内毒素进行直接比较得到的,内毒素的量用USP内毒素单位(USP-EU)表示。
[注:1 USP-EU等于1 IU 的内毒素]由于有专门制备的浊度法或显色法鲎试剂,因此也可按照要求使用这些检测方法。
这些试验方法要求先建立一条标准回归曲线,供试品的内毒素含量通过该曲线求得。
试验过程包括将内毒素和对照溶液与鲎试剂的反应管按照预先设定的时间恒温孵育,以及在适当波长用分光光度计读取吸光度。
在终点浊度法检测中,孵育期结束时应立刻读取读数;在终点显色法检测中,在到达预先设定的反应时间时,添加酶停止反应试剂使反应停止后再读取读数。
在动态浊度和显色法中,在整个反应期间始终对光密度的进行测定,而百分比值是通过这些读数来确定的。
器械和玻璃用具所有玻璃用具和耐热材料应置于热空气烤箱中有效除热原,♦2通常使用的最少时间和最低温度为30分钟,250℃。
如果使用塑料用具,如微孔板和自动加样器配套的吸头之类的,应确认其不含可测内毒素并不会干扰试验。
[注:在这一章中,“管”这个词泛指其它任何容器,如微量孔板的孔]♦1除了内毒素,鲎试剂也与某些β-葡聚糖反应,一些经过处理而制备的鲎试剂不会与β-葡聚糖反应,对于含有葡聚糖的样品的检测应用此类试剂。
ment can be a serious source of bias. In general, the rejection of measurements solely on the basis of their relative magnitudesis a procedure that should be used sparingly.Each suspected potency measurement, or outlier, may be tested against the following criterion. This criterion is based on the variation within a single group of supposedly equivalent measurements from a normal distribution. On average, it will reject a valid observation once in 25 trials or once in 50 trials. Designate the measurements in order of magnitude from y1to yN, wherey1is the candidate outlier, and N is the number of measurements in the group. Compute the relative gap by using Table A2-1, Test for Outlier Measurements, and the formulas below:When N = 3 to 7:G1= (y2− y1)/(yN− y1)When N = 8 to 10:G2= (y2− y1)/(yN − 1− y1)When N = 11 to 13:G3= (y3− y1)/(yN −1− y1)If G1, G2, or G3, as appropriate, exceeds the critical value in Table A2-1, Test for Outlier Measurements, for the observed N, there is a statistical basis for omitting the outlier measurement(s).Table A2-1. Test for Outlier MeasurementsIn samples from a normal population, gaps equal to or larger than the following values of G1, G2, and G3 occur with a probability P = 0.01, when outlier measurements can occur only at one end; or with P = 0.02, when they may occur at either end.N34567G10.9870.8890.7810.6980.637N8910G20.6810.6340.597N111213G30.6740.6430.617EXAMPLEEstimated potencies of sample in log scale = 1.561, 1.444, 1.517, 1.535.Check lowest potency for outlier:G1= (1.517 − 1.444)/(1.561 − 1.444) = 0.624<0.889Therefore 1.444 is not an outlier.Check highest potency for outlier:G1= (1.561 − 1.535)/(1.561 − 1.444) = 0.222<0.889Therefore 1.561 is not an outlier.Outlier potencies should be marked as outlier values and excluded from the assay calculations. NMT one potency can be excluded as an outlier.á85ñ BACTERIAL ENDOTOXINS TEST♦Portions of this general chapter have been harmonized with the corresponding texts of the European Pharmacopoeia and/or the Japanese Pharmacopoeia. Those portions that are not harmonized are marked with symbols (♦♦) to specify this fact.♦The Bacterial Endotoxins Test (BET) is a test to detect or quantify endotoxins from Gram-negative bacteria using amoebo-cyte lysate from the horseshoe crab (Limulus polyphemus or Tachypleus tridentatus).There are three techniques for this test: the gel-clot technique, which is based on gel formation; the turbidimetric technique, based on the development of turbidity after cleavage of an endogenous substrate; and the chromogenic technique, based on the development of color after cleavage of a synthetic peptide-chromogen complex. Proceed by any of the three techniques for the test. In the event of doubt or dispute, the final decision is made based upon the gel-clot limit test unless otherwise USP 40Biological Tests / á85ñ Bacterial Endotoxins Test 163indicated in the monograph for the product being tested. The test is carried out in a manner that avoids endotoxin contami-nation.APPARATUSDepyrogenate all glassware and other heat-stable materials in a hot air oven using a validated process.♦1♦ A commonly used minimum time and temperature is 30 min at 250°. If employing plastic apparatus, such as microplates and pipet tips for auto-matic pipetters, use apparatus that is shown to be free of detectable endotoxin and does not interfere in the test. [NOTE —In this chapter, the term “tube” includes any other receptacle such as a microtiter well.]REAGENTS AND TEST SOLUTIONSAmoebocyte LysateA lyophilized product obtained from the lysate of amoebocytes (white blood cells) from the horseshoe crab (Limulus polyphe-mus or Tachypleus tridentatus ). This reagent refers only to a product manufactured in accordance with the regulations of the competent authority. [NOTE —Amoebocyte Lysate reacts to some b -glucans in addition to endotoxins. Amoebocyte Lysate prepa-rations that do not react to glucans are available: they are prepared by removing the G factor reacting to glucans from Amoe-bocyte Lysate or by inhibiting the G factor reacting system of Amoebocyte Lysate and may be used for endotoxin testing in the presence of glucans.]Water for Bacterial Endotoxins Test (BET)Use Water for Injection or water produced by other procedures that shows no reaction with the lysate employed, at the de-tection limit of the reagent.Lysate TSDissolve Amoebocyte Lysate in Water for BET , or in a buffer recommended by the lysate manufacturer, by gentle stirring.Store the reconstituted lysate, refrigerated or frozen, according to the specifications of the manufacturer.PREPARATION OF SOLUTIONSStandard Endotoxin Stock SolutionA Standard Endotoxin Stock Solution is prepared from a USP Endotoxin Reference Standard that has been calibrated to the current WHO International Standard for Endotoxin. Follow the specifications in the package leaflet and on the label for prepa-ration and storage of the Standard Endotoxin Stock Solution . Endotoxin is expressed in Endotoxin Units (EU). [N OTE —One USP Endotoxin Unit (EU) is equal to one International Unit (IU) of endotoxin.]Standard Endotoxin SolutionsAfter mixing the Standard Endotoxin Stock Solution vigorously, prepare appropriate serial dilutions of Standard Endotoxin Solu-tion , using Water for BET . Use dilutions as soon as possible to avoid loss of activity by adsorption.Sample SolutionsPrepare the Sample Solutions by dissolving or diluting drugs using Water for BET . Some substances or preparations may be more appropriately dissolved, or diluted in other aqueous solutions. If necessary, adjust the pH of the solution to be examined (or dilution thereof) so that the pH of the mixture of the lysate and Sample Solution falls within the pH range specified by the lysate manufacturer, usually 6.0–8.0. The pH may be adjusted by use of an acid, base, or suitable buffer as recommended by the lysate manufacturer. Acids and bases may be prepared from concentrates or solids with Water for BET in containers free of detectable endotoxin. Buffers must be validated to be free of detectable endotoxin and interfering factors.♦1 For a validity test of the procedure for inactivating endotoxins, see Dry-Heat Sterilization under Sterilization and Sterility Assurance of Compendial Articles á1211ñ.Use Lysate TS having a sensitivity of not less than 0.15 Endotoxin Unit per mL.♦164 á85ñ Bacterial Endotoxins Test / Biological Tests USP 40DETERMINATION OF MAXIMUM VALID DILUTION (MVD)The maximum valid dilution is the maximum allowable dilution of a specimen at which the endotoxin limit can be deter-mined. Determine the MVD from the following equation:MVD = (endotoxin limit × concentration of Sample Solution)/(l)Endotoxin LimitThe endotoxin limit for parenteral drugs, defined on the basis of dose, equals K/M♦2♦, where K is a threshold pyrogenic dose of endotoxin per kg of body weight, and M is equal to the maximum recommended bolus dose of product per kg of body weight. When the product is to be injected at frequent intervals or infused continuously, M is the maximum total dose admin-istered in a single hour period. The endotoxin limit for parenteral drugs is specified in the individual monograph in units such as EU/mL, EU/mg, EU/Unit of biological activity, etc.Concentration of Sample Solutionmg/mL: in the case of endotoxin limit specified by weight (EU/mg);Units/mL: in the case of endotoxin limit specified by unit of biological activity (EU/Unit);mL/mL: when the endotoxin limit is specified by volume (EU/mL).l: the labeled sensitivity in the Gel-Clot Technique (EU/mL) or the lowest concentration used in the standard curve for the Turbidimetric Technique or Chromogenic Technique.GEL-CLOT TECHNIQUEThe gel-clot technique is used for detecting or quantifying endotoxins based on clotting of the lysate reagent in the pres-ence of endotoxin. The minimum concentration of endotoxin required to cause the lysate to clot under standard conditions is the labeled sensitivity of the lysate reagent. To ensure both the precision and validity of the test, perform the tests for confirm-ing the labeled lysate sensitivity and for interfering factors as described in Preparatory Testing, immediately below.Preparatory TestingTEST FOR CONFIRMATION OF LABELED LYSATE SENSITIVITYConfirm in four replicates the labeled sensitivity, l, expressed in EU/mL of the lysate prior to use in the test. The test for confirmation of lysate sensitivity is to be carried out when a new batch of lysate is used or when there is any change in the test conditions that may affect the outcome of the test. Prepare standard solutions having at least four concentrations equivalent to 2l, l, 0.5l, and 0.25l by diluting the USP Endotoxin RS with Water for BET.Mix a volume of the Lysate TS with an equal volume (such as 0.1-mL aliquots) of one of the Standard Endotoxin Solutions in each test tube. When single test vials or ampuls containing lyophilized lysate are used, add solutions directly to the vial or am-pul. Incubate the reaction mixture for a constant period according to the directions of the lysate manufacturer (usually at37±1° for 60±2 min), avoiding vibration. To test the integrity of the gel, take each tube in turn directly from the incubator, and invert it through about 180° in one smooth motion. If a firm gel has formed that remains in place upon inversion, record the result as positive. A result is negative if an intact gel is not formed. The test is considered valid when the lowest concentra-tion of the standard solutions shows a negative result in all replicate tests.The endpoint is the smallest concentration in the series of decreasing concentrations of standard endotoxin that clots the lysate. Determine the geometric mean endpoint by calculating the mean of the logarithms of the endpoint concentrations of the four replicate series and then taking the antilogarithm of the mean value, as indicated in the following formula:geometric mean endpoint concentration = antilog (S e/f)where S e is the sum of the log endpoint concentrations of the dilution series used, and f is the number of replicate test tubes. The geometric mean endpoint concentration is the measured sensitivity of the lysate (in EU/mL). If this is not less than 0.5l and not more than 2l, the labeled sensitivity is confirmed and is used in tests performed with this lysate.♦2K is 5 USP-EU/kg of body weight for any route of administration other than intrathecal (for which K is 0.2 USP-EU/kg of body weight). For radiopharmaceutical products not administered intrathecally, the endotoxin limit is calculated as 175 EU/V, where V is the maximum recommended dose in mL. For intrathecally ad-ministered radiopharmaceuticals, the endotoxin limit is obtained by the formula 14 EU/V. For formulations (usually anticancer products) administered on a per square meter of body surface, the formula is K/M, where K = 100 EU/m2 and M is the maximum dose/m2.♦USP 40Biological Tests / á85ñ Bacterial Endotoxins Test 165TEST FOR INTERFERING FACTORSUsually prepare solutions (A–D) as shown in Table 1, and perform the inhibition/enhancement test on the Sample Solutions at a dilution less than the MVD, not containing any detectable endotoxins, operating as described for Test for Confirmation of Labeled Lysate Sensitivity . The geometric mean endpoint concentrations of Solutions B and C are determined using the formula described in the Test for Confirmation of Labeled Lysate Sensitivity . The test for interfering factors must be repeated when any condition changes that is likely to influence the result of the test.Table 1. Preparation of Solutions for the Inhibition/Enhancement Test for Gel-Clot TechniquesSolution Endotoxin Concentration/Solution to Which Endotoxin Is Added Diluent Dilution Factor Endotoxin Concentration NumberofReplicates A a None/Sample Solution ———4B b 2l /Sample Solution Sample Solution 12l 4 21l 4 40.5l 4 80.25l 4C c 2l /Water for BET Water for BET 12l 2 21l 240.5l 280.25l 2D d None/Water for BET ———2a Solution A : A Sample Solution of the preparation under test that is free of detectable endotoxins.bSolution B : Test for interference.c Solution C : Control for labeled lysate sensitivity.dSolution D : Negative control of Water for BET.The test is considered valid when all replicates of Solutions A and D show no reaction and the result of Solution C confirmsthe labeled sensitivity.If the sensitivity of the lysate determined in the presence of Solution B is not less than 0.5l and not greater than 2l , theSample Solution does not contain factors that interfere under the experimental conditions used. Otherwise, the Sample Solution to be examined interferes with the test.If the sample under test does not comply with the test at a dilution less than the MVD, repeat the test using a greater dilu-tion, not exceeding the MVD. The use of a more sensitive lysate permits a greater dilution of the sample to be examined, and this may contribute to the elimination of interference.Interference may be overcome by suitable treatment such as filtration, neutralization, dialysis, or heating. To establish that the chosen treatment effectively eliminates interference without loss of endotoxins, perform the assay described above using the preparation to be examined to which Standard Endotoxin has been added and which has then been submitted to the chosen treatment.Limit TestPROCEDUREPrepare Solutions A, B, C, and D as shown in Table 2, and perform the test on these solutions following the procedure above for Preparatory Testing , Test for Confirmation of Labeled Lysate Sensitivity .Table 2. Preparation of Solutions for the Gel-Clot Limit TestSolution *Endotoxin Concentration/Solution to WhichEndotoxin Is Added Number of ReplicatesA None/Diluted Sample Solution 2B 2l /Diluted Sample Solution 2C 2l /Water for BET 2D None/Water for BET 2* Prepare Solution A and the positive product control Solution B using a dilution not greater than the MVD and treatments as described for the Test for Interfering Factors in Preparatory Testing . The positive control Solutions B and C contain the Standard Endotoxin Solution at a concentration corresponding to twice the la-beled lysate sensitivity. The negative control Solution D consists of Water for BET.166 á85ñ Bacterial Endotoxins Test / Biological Tests USP 40INTERPRETATIONThe test is considered valid when both replicates of Solutions B and C are positive and those of Solution D are negative. When a negative result is found for both replicates of Solution A, the preparation under test complies with the test. When a positive result is found for both replicates of Solution A, the preparation under test does not comply with the test.When a positive result is found for one replicate of Solution A and a negative result is found for the other, repeat the test. In the repeat test, the preparation under test complies with the test if a negative result is found for both replicates of Solution A. The preparation does not comply with the test if a positive result is found for one or both replicates of Solution A. However, if the preparation does not comply with the test at a dilution less than the MVD, the test may be repeated using a greater dilu-tion, not exceeding the MVD.Quantitative TestPROCEDUREThe test quantifies bacterial endotoxins in Sample Solutions by titration to an endpoint. Prepare Solutions A, B, C, and D as shown in Table 3, and test these solutions by following the procedure in Preparatory Testing, Test for Confirmation of Labeled Lysate Sensitivity.Table 3. Preparation of Solutions for the Gel-Clot AssaySolutionEndotoxin Concentration/Solution to Which EndotoxinIs Added DiluentDilutionFactorEndotoxinConcentrationNumberofReplicatesA a None/Sample Solution Water for BET1—22—24—28—2B b2l/Sample Solution—12l2C c2l/Water for BET Water for BET12l221l240.5l280.25l2D d None/Water for BET———2a Solution A: Sample Solution under test at the dilution, not to exceed the MVD, with which the Test for Interfering Factors was completed. Subsequent dilution of the Sample Solution must not exceed the MVD. Use Water for BET to make a dilution series of four tubes containing the Sample Solution under test at concentra-tions of 1, 1/2, 1/4, and 1/8 relative to the concentration used in the Test for Interfering Factors. Other dilutions up to the MVD may be used as appropriate.b Solution B: Solution A containing standard endotoxin at a concentration of 2l (positive product control).c Solution C: Two replicates of four tubes of Water for BET containing the standard endotoxin at concentrations of 2l, l, 0.5l, and 0.25l, respectively.d Solution D: Water for BET (negative control).CALCULATION AND INTERPRETATIONThe test is considered valid when the following three conditions are met: (1) Both replicates of negative control Solution D are negative; (2) Both replicates of positive product control Solution B are positive; and (3) The geometric mean endpoint con-centration of Solution C is in the range of 0.5l to 2l.To determine the endotoxin concentration of Solution A, calculate the endpoint concentration for each replicate by multiply-ing each endpoint dilution factor by l. The endotoxin concentration in the Sample Solution is the endpoint concentration of the replicates. If the test is conducted with a diluted Sample Solution, calculate the concentration of endotoxin in the original Sample Solution by multiplying by the dilution factor. If none of the dilutions of the Sample Solution is positive in a valid assay, report the endotoxin concentration as less than l (if the diluted sample was tested, report as less than l times the lowest dilu-tion factor of the sample). If all dilutions are positive, the endotoxin concentration is reported as equal to or greater than the greatest dilution factor multiplied by l (e.g., initial dilution factor times eight times l in Table 3).The preparation under test meets the requirements of the test if the concentration of endotoxin in both replicates is less than that specified in the individual monograph.USP 40Biological Tests / á85ñ Bacterial Endotoxins Test 167PHOTOMETRIC QUANTITATIVE TECHNIQUESTurbidimetric TechniqueThis technique is a photometric assay measuring increases in reactant turbidity. On the basis of the particular assay principle employed, this technique may be classified as either an endpoint-turbidimetric assay or a kinetic-turbidimetric assay. The end-point-turbidimetric assay is based on the quantitative relationship between the concentration of endotoxins and the turbidity (absorbance or transmission) of the reaction mixture at the end of an incubation period. The kinetic-turbidimetric assay is a method to measure either the time (onset time) needed to reach a predetermined absorbance or transmission of the reaction mixture, or the rate of turbidity development. The test is carried out at the incubation temperature recommended by the ly-sate manufacturer (which is usually 37±1°).Chromogenic TechniqueThis technique is an assay to measure the chromophore released from a suitable chromogenic peptide by the reaction of endotoxins with lysate. On the basis of the particular assay principle employed, this technique may be classified as either an endpoint-chromogenic assay or a kinetic-chromogenic assay. The endpoint-chromogenic assay is based on the quantitative re-lationship between the concentration of endotoxins and the release of chromophore at the end of an incubation period. The kinetic-chromogenic assay is a method to measure either the time (onset time) needed to reach a predetermined absorbance of the reaction mixture, or the rate of color development. The test is carried out at the incubation temperature recommendedby the lysate manufacturer (which is usually 37±1°).Preparatory Testing To assure the precision or validity of the turbidimetric and chromogenic techniques, preparatory tests are conducted to veri-fy that the criteria for the standard curve are valid and that the sample solution does not interfere with the test. Validation for the test method is required when conditions that are likely to influence the test result change.ASSURANCE OF CRITERIA FOR THE STANDARD CURVEThe test must be carried out for each lot of lysate reagent. Using the Standard Endotoxin Solution, prepare at least three en-dotoxin concentrations within the range indicated by the lysate manufacturer to generate the standard curve. Perform the as-say using at least three replicates of each standard endotoxin concentration according to the manufacturer's instructions for the lysate (volume ratios, incubation time, temperature, pH, etc.). If the desired range is greater than two logs in the kinetic methods, additional standards should be included to bracket each log increase in the range of the standard curve. The abso-lute value of the correlation coefficient, r , must be greater than or equal to 0.980 for the range of endotoxin concentrations set up.TEST FOR INTERFERING FACTORSSelect an endotoxin concentration at or near the middle of the endotoxin standard curve. Prepare Solutions A, B, C, and D as shown in Table 4. Perform the test on Solutions A, B, C, and D at least in duplicate, according to the instructions for the lysate employed, for example, concerning volume of Sample Solution and Lysate TS, volume ratio of Sample Solution to Lysate TS,incubation time, etc.Table 4. Preparation of Solutions for the Inhibition/Enhancement Test for Photometric TechniquesSolutionEndotoxin Concentration Solution to WhichEndotoxin Is Added Number of Replicates A a None Sample Solution Not less than 2B b Middle concentration of the standard curve Sample Solution Not less than 2C c At least three concentrations (lowest concentration is des-ignated l )Water for BET Each not less than 2D d None Water for BET Not less than 2a Solution A : The Sample Solution may be diluted not to exceed MVD.b Solution B : The preparation under test at the same dilution as Solution A, containing added endotoxin at a concentration equal to or near the middle of the standard curve.c Solution C : The standard endotoxin at the concentrations used in the validation of the method described for Assurance of Criteria for the Standard Curve under Preparatory Testing (positive controls).d Solution D : Water for BET (negative control).The test is considered valid when the following conditions are met.168 á85ñ Bacterial Endotoxins Test / Biological Tests USP 401.The absolute value of the correlation coefficient of the standard curve generated using Solution C is greater than or equalto 0.980.2.The result with Solution D does not exceed the limit of the blank value required in the description of the lysate reagentemployed, or it is less than the endotoxin detection limit of the lysate reagent employed.Calculate the mean recovery of the added endotoxin by subtracting the mean endotoxin concentration in the solution, if any (Solution A, Table 4), from that containing the added endotoxin (Solution B, Table 4). In order to be considered free of factors that interfere with the assay under the conditions of the test, the measured concentration of the endotoxin added to the Sample Solution must be within 50%–200% of the known added endotoxin concentration after subtraction of any endo-toxin detected in the solution without added endotoxin.When the endotoxin recovery is out of the specified range, the Sample Solution under test is considered to contain interfer-ing factors. Then, repeat the test using a greater dilution, not exceeding the MVD. Furthermore, interference of the Sample Solution or diluted Sample Solution not to exceed the MVD may be eliminated by suitable validated treatment such as filtration, neutralization, dialysis, or heat treatment. To establish that the chosen treatment effectively eliminates interference without loss of endotoxins, perform the assay described above, using the preparation to be examined to which Standard Endotoxin has been added and which has then been submitted to the chosen treatment.Test ProcedureFollow the procedure described for Test for Interfering Factors under Preparatory Testing, immediately above.CalculationCalculate the endotoxin concentration of each of the replicates of Solution A, using the standard curve generated by the positive control Solution C. The test is considered valid when the following three requirements are met.1.The results of the control Solution C comply with the requirements for validation defined for Assurance of Criteria for theStandard Curve under Preparatory Testing.2.The endotoxin recovery, calculated from the concentration found in Solution B after subtracting the concentration of en-dotoxin found in Solution A, is within the range of 50%–200%.3.The result of the negative control Solution D does not exceed the limit of the blank value required in the description of thelysate employed, or it is less than the endotoxin detection limit of the lysate reagent employed.InterpretationIn photometric assays, the preparation under test complies with the test if the mean endotoxin concentration of the repli-cates of Solution A, after correction for dilution and concentration, is less than the endotoxin limit for the product.á87ñ BIOLOGICAL REACTIVITY TESTS, IN VITROThe following tests are designed to determine the biological reactivity of mammalian cell cultures following contact with the elastomeric plastics and other polymeric materials with direct or indirect patient contact or of specific extracts prepared from the materials under test. It is essential that the tests be performed on the specified surface area. When the surface area of the specimen cannot be determined, use 0.1 g of elastomer or 0.2 g of plastic or other material for every mL of extraction fluid. Exercise care in the preparation of the materials to prevent contamination with microorganisms and other foreign matter. Three tests are described (i.e., the Agar Diffusion Test, the Direct Contact Test, and the Elution Test).1 The decision as to which type of test or the number of tests to be performed to assess the potential biological response of a specific sample or extract depends upon the material, the final product, and its intended use. Other factors that may also affect the suitability of a sample for a specific use are the polymeric composition; processing and cleaning procedures; contacting media; inks; adhe-sives; absorption, adsorption, and permeability of preservatives; and conditions of storage. Evaluation of such factors should be made by appropriate additional specific tests before determining that a product made from a specific material is suitable for its intended use. Materials that fail the in vitro tests are candidates for the in vivo tests described in Biological Reactivity Tests, In Vivo á88ñ.1 Further details are given in the following publications of the American Society for Testing and Materials, 1916 Race St., Philadelphia, PA 19103: Standard test method for agar diffusion cell culture screening for cytotoxicity, ASTM Designation F 895-84; Standard practice for direct contact cell culture evaluation of materi-als for medical devices, ASTM Designation F 813-83.USP 40Biological Tests / á87ñ Biological Reactivity Tests, In Vitro 169。
REAGENTS AND TEST SOLUTIONS 试剂和测试溶液Amoebocyte Lysate— A lyophilized product obtained from the lysate of amoebocytes (white blood cells) from the horseshoe crab (Limulus polyphemus or Tachypleus tridentatus). This reagent refers only to a product manufactured in accordance with the regulations of the competent authority. [NOTE—Amoebocyte Lysate reacts to some-glucans in addition to endotoxins. Amoebocyte Lysate preparations that do not react to glucans are available: they are prepared by removing the G factor reacting to glucans from Amoebocyte Lysate or by inhibiting the G factor reacting system of Amoebocyte Lysate and may be used for endotoxin testing in the presence of glucans. ] 变形细胞溶解液—由鲎的〔白血细胞〕变形细胞溶解液制得的冻干产品。
该试剂仅指那些按照相关的监管机构的法规要求制得的产品。
[注:变形细胞溶解液除了与内毒素反响外,还会与某些-葡聚糖反响。
可以制成不与-葡聚糖发生反响的变形细胞溶解液制品:可通过从变形细胞溶解液中去除会与葡聚糖反响的G因子或抑制变形细胞溶解液的G因子反响系统来制得该制品,这些制品可用于在存在葡聚糖时进行内毒素检测。
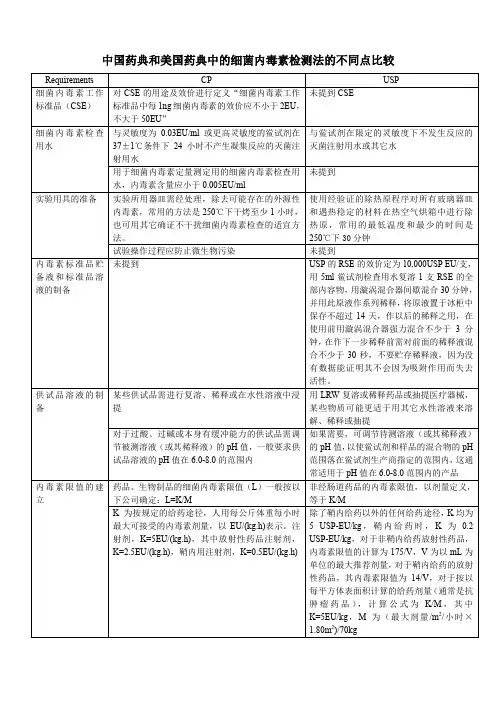
中国药典和美国药典中的细菌内毒素检测法的不同点比较1、细菌内毒素工作标准品(CSE)CP 对CSE的用途及效价进行定义“细菌内毒素工作标准品中每1ng细菌内毒素的效价应不小于2EU,不大于50EU”USP未提到CSE2、细菌内毒素检查用水CP :与灵敏度为0.03EU/ml或更高灵敏度的鲎试剂在37±1℃条件下24小时不产生凝集反应的灭菌注射用水;USP:与鲎试剂在限定的灵敏度下不发生反应的灭菌注射用水或其它水。
CP:用于细菌内毒素定量测定用的细菌内毒素检查用水,内毒素含量应小于0.005EU/ml。
USP:未提到。
3、实验用具的准备CP:实验所用器皿需经处理,除去可能存在的外源性内毒素,常用的方法是250℃下干烤至少1小时,也可用其它确证不干扰细菌内毒素检查的适宜方法。
USP:使用经验证的除热原程序对所有玻璃器皿和遇热稳定的材料在热空气烘箱中进行除热原,常用的最低温度和最少的时间是250℃下30分钟。
4、内毒素标准品贮备液和标准品溶液的制备CP:未提到USP:RSE的效价定为10,000USP EU/支,用5ml鲎试剂检查用水复溶1支RSE的全部内容物,用漩涡混合器间歇混合30分钟,并用此原液作系列稀释,将原液置于冰柜中保存不超过14天,作以后的稀释之用,在使用前用漩涡混合器强力混合不少于3分钟,在作下一步稀释前需对前面的稀释液混合不少于30秒,不要贮存稀释液,因为没有数据能证明其不会因为吸附作用而失去活性。
5、供试品溶液的制备CP:|某些供试品需进行复溶、稀释或在水性溶液中浸提用LRW复溶或稀释药品或抽提医疗器械,某些物质可能更适于用其它水性溶液来溶解、稀释或抽提。
对于过酸、过碱或本身有缓冲能力的供试品需调节被测溶液(或其稀释液)的pH值,一般要求供试品溶液的pH值在6.0-8.0的范围内。
USP:用LRW复溶或稀释药品或抽提医疗器械,某些物质可能更适于用其它水性溶液来溶解、稀释或抽提。
BACTERIAL ENDOTOXINS TEST <85> USP35Portions of this general chapter have been harmonized with the corresponding texts ofthe European Pharmacopoeia and/or the Japanese Pharmacopoeia. Those portions that are not harmonized are marked with symbols () to specify this fact.这一章节已经和欧洲药典(EP)与日本药典(JP)统一。
没有统一的部分已经用标记出来。
The Bacterial Endotoxins Test (BET) is a test to detect or quantify endotoxins from Gram-negative bacteria using amoebocyte lysate from the horseshoe crab (Limulus polyphemus or Tachypleus tridentatus).细菌内毒素检查法(BET)是通过鲎的阿米巴细胞溶解产物来检测或定量来自革兰氏阴性菌的内毒素。
There are three techniques for this test: the gel-clot technique, which is based on gel formation; the turbidimetric technique, based on the development of turbidity after cleavage of an endogenous substrate; and the chromogenic technique, based on the development of color after cleavage of a synthetic peptide-chromogen complex. Proceed by any of the three techniques for the test. In the event of doubt or dispute, the final decision is made based upon the gel-clot technique unless otherwise indicated in the monograph for the product being tested. The test is carried out in a manner that avoids endotoxin contamination.细菌内毒素检查法有3种:凝胶法,基于凝胶的形成;浊度法,BACTERIAL ENDOTOXINS TEST USP32Portions of this general chapter have been harmonized with the corresponding texts of the European Pharmacopeia and/or the Japanese Pharmacopeia. Those portions that are notharmonized are marked with symbols () to specify this fact.This chapter provides a test to detect or quantify bacterial endotoxins that may be present in or on the sample of the article(s) to which the test is applied. It uses Limulus Amebocyte Lysate (LAL) obtained from the aqueous extracts of circulating amebocytes of horseshoe crab (Limulus polyphemus or Tachypleus tridentatus) which has been prepared and characterized for use as anLAL Reagent.1There are two types of techniques for this test: the gel-clot techniques, which are based on gel formation, and the photometric techniques. The latter include a turbidimetric method, which is based on the development of turbidity after cleavage of an endogenous substrate, and a chromogenic method, which is based on the development of color after cleavage of a synthetic peptide-chromogen complex. Proceed by any one of these techniques, unless otherwise indicated in the monograph. In case of dispute, the final decision is based on the gel-clot techniques, unless otherwise indicated in the monograph.In the gel-clot techniques, the reaction endpoint is determined from dilutions of the material under test in direct comparison with parallel dilutions of a reference endotoxin, and quantities of endotoxin are expressed in USP Endotoxin Units (USP-EU).[NOTE—One USP-EU is equal to one IU of endotoxin.]Because LAL Reagents have been formulated to be used also for turbidimetric or colorimetric tests, such tests may be used to comply with the requirements. These tests require the establishment of a standard regression curve; the endotoxin content of the test material is determined by interpolation from the curve. The procedures include incubation for a preselected time of reacting endotoxin and control solutions with LAL Reagent and reading of the spectrophotometric light absorbance at suitable wavelengths. In the endpoint turbidimetric procedure the reading is made immediately at the end of the incubation period. In the endpoint colorimetric procedure the reaction is arrested at the end of the preselected time by the addition of an enzyme reaction-terminating agent prior to the readings. In the turbidimetric and colorimetric kinetic assays the absorbance is measured throughout the reaction period and rate values are determined from those readings.APPARATUS AND GLASSWAREDepyrogenate all glassware and other heat-stable materials in a hot-air oven using a validatedprocess.2Commonly used minimum time and temperature settings are 30 minutes at 250.If employing plastic apparatus, such as microplates and pipet tips for automatic pipetters, use only that which has been shown to be free of detectable endotoxin and not to interfere with thetest.[NOTE—In this chapter, the term “tube” includes any other receptacle such as a micro-titer well.]PREPARATION OF THE STANDARD ENDOTOXIN STOCK SOLUTION AND STANDARD SOLUTIONS The USP Endotoxin RS has a defined potency of 10,000 USP Endotoxin Units (EU) per vial. Constitute the entire contents of 1 vial of the RSE with 5 mL of LAL Reagent Water3, mixintermittently for 30 minutes, using a vortex mixer, and use this concentrate for making appropriate serial dilutions. Preserve the concentrate in a refrigerator for making subsequent dilutions for not more than 14 days. Mix vigorously, using a vortex mixer, for not less than 3 minutes before use. Mix each dilution for not less than 30 seconds before proceeding to make the next dilution. Do not store dilutions, because of loss of activity by adsorption, in the absence of supporting data to the contrary.Preparatory TestingUse an LAL Reagent of confirmed label sensitivity.The validity of test results for bacterial endotoxins requires an adequate demonstration that specimens of the article or of solutions, washings, or extracts thereof to which the test is to be applied do not of themselves inhibit or enhance the reaction or otherwise interfere with the test. Validation is accomplished by performing the inhibition or enhancement test described under each of the three techniques indicated. Appropriate negative controls are included. Validation must be repeated if the LAL Reagent source or the method of manufacture or formulation of the article is changed.Preparation of Sample SolutionsPrepare sample solutions by dissolving or diluting drugs or extracting medical devices using LAL Reagent Water. Some substances or preparations may be more appropriately dissolved, diluted, or extracted in other aqueous solutions. If necessary, adjust the pH of the solution (or dilution thereof) to be examined so that the pH of the mixture of the LAL Reagent and sample falls within the pH range specified by the LAL Reagent manufacturer. This usually applies to a product with a pH in the range of 6.0 to 8.0. The pH may be adjusted using an acid, base, or suitable buffer as recommended by the LAL Reagent manufacturer. Acids and bases may be prepared from concentrates or solids with LAL Reagent Water in containers free of detectable endotoxin. Buffers must be validated to be free of detectable endotoxin and interfering factors.DETERMINATION OF MAXIMUM VALID DILUTION (MVD)The Maximum Valid Dilution is the maximum allowable dilution of a specimen at which the endotoxin limit can be determined. It applies to injections or to solutions for parenteral administration in the form constituted or diluted for administration, or, where applicable, to the amount of drug by weight if the volume of the dosage form for administration could be varied. The general equation to determine MVD is:MVD = (Endotoxin limit × Concentration of sample solution)/()where the concentration of sample solution and are as defined below. Where the endotoxinlimit concentration is specified in the individual monograph in terms of volume (in EU per mL), divide the limit by, which is the labeled sensitivity (in EU per mL) of the LAL Reagent, to obtainthe MVD factor. Where the endotoxin limit concentration is specified in the individual monograph in terms of weight or Units of active drug (in EU per mg or in EU per Unit), multiply the limit by the concentration (in mg per mL or in Units per mL) of the drug in the solution tested or of the drug constituted according to the label instructions, whichever is applicable, and dividethe product of the multiplication by, to obtain the MVD factor. The MVD factor so obtained isthe limit dilution factor for the preparation for the test to be valid.ESTABLISHMENT OF ENDOTOXIN LIMITSThe endotoxin limit for parenteral drugs, defined on the basis of dose, is equal to K/M,4where K is the threshold human pyrogenic dose of endotoxin per kg of body weight,and M is equal to the maximum recommended human dose of product per kg of body weight in a single hour period.The endotoxin limit for parenteral drugs is specified in individual monographs in units such as EU/mL, EU/mg, or EU/Unit of biological activity.GEL-CLOT TECHNIQUESThe gel-clot techniques detect or quantify endotoxins based on clotting of the LAL Reagent in the presence of endotoxin. The concentration of endotoxin required to cause the lysate to clot under standard conditions is the labeled sensitivity of the LAL Reagent. To ensure both the precision and validity of the test, tests for confirming the labeled LAL Reagent sensitivity and for interfering factors are described under Preparatory Testing for the Gel-Clot Techniques.Preparatory Testing for the Gel-Clot TechniquesTest for Confirmation of Labeled LAL Reagent Sensitivity—Confirm the labeled sensitivity using at least 1 vial of the LAL Reagent lot. Prepare a series of two-fold dilutions of the USP EndotoxinRS in LAL Reagent Water to give concentrations of 2,, 0.5, and 0.25, where is asdefined above. Perform the test on the four standard concentrations in quadruplicate and include negative controls. The test for confirmation of lysate sensitivity is to be carried out when a new batch of LAL Reagent is used or when there is any change in the experimental conditions that may affect the outcome of the test.Mix a volume of the LAL Reagent with an equal volume (such as 0.1-mL aliquots) of one of the standard solutions in each test tube. When single test vials or ampuls containing lyophilized LAL Reagent are used, add solutions directly to the vial or ampul. Incubate the reaction mixture for aconstant period according to directions of the LAL Reagent manufacturer (usually at 37 ± 1for 60 ± 2 minutes), avoiding vibration. To test the integrity of the gel, take each tube in turn directly from the incubator and invert it through about 180in one smooth motion. If a firm gel hasformed that remains in place upon inversion, record the result as positive. A result is negative if an intact gel is not formed. The test is not valid unless the lowest concentration of the standard solutions shows a negative result in all replicate tests.The endpoint is the last positive test in the series of decreasing concentrations of endotoxin. Calculate the mean value of the logarithms of the endpoint concentration and then the antilogarithm of the mean value using the following equation:Geometric Mean Endpoint Concentration = antilog (S e / f)where S e is the sum of the log endpoint concentrations of the dilution series used, and f is the number of replicate test tubes. The geometric mean endpoint concentration is the measuredsensitivity of the LAL Reagent (in EU/mL). If this is not less than 0.5and not more than 2, the labeled sensitivity is confirmed and is used in tests performed with this lysate.Interfering Factors Test for the Gel-Clot Techniques—Prepare solutions A, B, C, and D as shown in Table 1, and perform the inhibition/enhancement test on the sample solutions at a dilution less than the MVD, not containing any detectable endotoxins, following the procedure inthe Test for Confirmation of Labeled LAL Reagent Sensitivity above. The geometric mean endpoint concentrations of solutions B and C are determined using the equation in that test.Table 1. Preparation of Solutions for the Inhibition/Enhancement Test for Gel-Clot Techniques2/sample solution 210.50.252/water for BET 210.50.25This test must be repeated when any condition that is likely to influence the test results changes. The test is not valid unless Solutions A and D show no reaction and the result of Solution C confirms the labeled sensitivity.If the sensitivity of the lysate determined in the presence of the sample solution under test of Solution B is not less than 0.5and not greater than 2, the sample solution does not containfactors which interfere under the experimental conditions used. Otherwise, the sample solution to be examined interferes with the test.If the sample under test does not comply with the test at a dilution less than the MVD, repeat the test using a greater dilution, not exceeding the MVD. The use of a more sensitive lysate permits a greater dilution of the sample to be examined and this may contribute to the elimination of interference.Interference may be overcome by suitable treatment, such as filtration, neutralization, dialysis, or heating. To establish that the chosen treatment effectively eliminates interference without loss of endotoxins, perform the assay described below using the preparation to be examined to which USP Endotoxin RS has been added and which has been subjected to the selected treatment.Gel-Clot Limit TestThis test is used when a monograph contains a requirement for endotoxin limits.Procedure—Prepare Solutions A, B, C, and D as shown in Table 2, and perform the test on these solutions following the procedure in the Test for Confirmation of Labeled LAL Reagent Sensitivity under Preparatory Testing for the Gel-Clot Techniques.Table 2. Preparation of Solutions for the Gel-Clot Limit Test2/diluted sample solution2/LAL Reagent WaterInterpretation—The test is not valid unless both replicates of positive control Solutions B and C are positive and those of negative control Solution D are negative. The preparation under test complies with the test when a negative result is found for both tubes containing Solution A. The preparation under test does not comply with the test when a positive result is found for both tubes containing Solution A.Repeat the test when a positive result is found for 1 tube containing Solution A and a negative result for the other one. The preparation under test complies with the test when a negative result is found for both tubes containing Solution A in the repeat result. If the test is positive for the preparation under test at a dilution less than the MVD, the test may be repeated at a dilution not greater than the MVD.Gel-Clot AssayThis assay quantifies bacterial endotoxins in sample solutions by titration to an endpoint.Procedure—Prepare Solutions A, B, C, and D as shown in Table 3, and test these solutions by following the procedure in the Test for Confirmation of Labeled LAL ReagentSensitivity under Preparatory Testing for the Gel-Clot Techniques.Table 3. Preparation of Solutions for the Gel-Clot Assay2/sample solution 22/LAL Reagent Water 210.50.25b Solution B: Solution A containing standard endotoxin at a concentration of 2(positive product control).c Solution C: two series of 4 tubes of LAL Reagent Water containing the standard endotoxin at a concentration of 2,, 0.5, and 0.25, respectively.Calculation and Interpretation—The test is not valid unless the following conditions are met: (1) both replicates of negative control Solution D are negative; (2) both replicates of positive product control Solution B are positive; and (3) the geometric mean endpoint concentration of Solution Cis in the range of 0.5to 2.To determine the endotoxin concentration of Solution A, calculate the endpoint concentration for each replicate series of dilutions by multiplying each endpoint dilution factor by. Theendotoxin concentration in the sample is the geometric mean endpoint concentration of the replicates (see the formula given in the Test for Confirmation of Labeled LAL Reagent Sensitivity under Preparatory Testing for the Gel-Clot Techniques). If the test is conducted with a diluted sample solution, calculate the concentration of endotoxin in the original sample solution by multiplying by the dilution factor. If none of the dilutions of the sample solution is positive in avalid assay, report the endotoxin concentration as less than(if the diluted sample was tested,less than times the lowest dilution factor of the sample.) If all dilutions are positive, the endotoxin concentration is reported as equal to or greater than the greatest dilution factor multiplied by(e.g., initial dilution factor times 8 times in Table 3).The article meets the requirements of the test if the concentration of endotoxin is less than that specified in the individual monograph.PHOTOMETRIC TECHNIQUESThe turbidimetric method measures increases in turbidity. Depending on the test principle used, this technique is classified as either endpoint-turbidimetric or kinetic-turbidimetric. The endpoint-turbidimetric technique is based on the quantitative relationship between the concentration of endotoxins and the turbidity (absorbance or transmission) of the reaction mixture at the end of an incubation period. The kinetic-turbidimetric technique is a method to measure either the onset time needed to reach a predetermined absorbance of the reaction mixture or the rate of turbidity development.The chromogenic method measures the chromophore released from a suitable chromogenic peptide by the reaction of endotoxins with the LAL Reagent. Depending on the test principle employed, this technique is classified as either endpoint-chromogenic or kinetic-chromogenic. The endpoint-chromogenic technique is based on the quantitative relationship between the concentration of endotoxins and the release of chromophore at the end of an incubation period. The kinetic-chromogenic technique is a method to measure either the onset time needed to reach a predetermined absorbance of the reaction mixture or the rate of color development.All photometric tests are carried out at the incubation temperature recommended by the LAL Reagent manufacturer, which is usually 37 ± 1.Preparatory Testing for the Photometric TechniquesTo assure the precision or validity of the turbidimetric and chromogenic techniques, preparatory tests are conducted to verify that the criteria for the standard curve are valid and that the sample solution does not inhibit or enhance the reaction. Revalidation for the test method is required when conditions that are likely to influence the test result change.Verification of Criteria for the Standard Curve—Using the Standard Endotoxin Solution, prepare at least three endotoxin concentrations to generate the standard curve. Perform the test using at least three replicates of each standard endotoxin concentration according to the manufacturer's instructions for the LAL Reagent (with regard to volume ratios, incubation time, temperature, pH, etc.). If the desired range in the kinetic methods is greater than two logs, additional standards should be included to bracket each log increase within the range of the standard curve. The absolute value of the correlation coefficient, |r|, must be greater than or equal to 0.980 for the range of endotoxin concentrations indicated by the manufacturer of the LAL Reagent.Interfering Factors Test for the Photometric Techniques—Select an endotoxin concentration at or near the middle of the endotoxin standard curve. Prepare Solutions A, B, C, and D as shownin Table 4. Perform the test on Solutions A, B, C, and D at least in duplicate following the instructions for the LAL Reagent used (with regard to volume of sample and LAL Reagent, volume ratio of sample to LAL Reagent, incubation time, etc.).Table 4. Preparation of Solutions for the Inhibition/Enhancement Test for PhotometricTechniquesat least 3 concentrations (lowestconcentration is designated)Calculate the mean recovery of the added endotoxin by subtracting the mean endotoxin concentration in the solution (if any) from that containing the added endotoxin. In order to be considered free of interfering factors under the conditions of the test, the measured concentration of the endotoxin added to the sample solution must be within 50% to 200% of the known added endotoxin concentration after subtraction of any endotoxin detected in the solution without added endotoxin.When the endotoxin recovery is out of the specified ranges, the interfering factors must be removed as described in the Interfering Factors Test for the Gel-ClotTechniques under Preparatory Testing for the Gel-Clot Techniques.Repeating the Interfering Factors Test for the Gel-Clot Techniques validates the treatment.Procedure for the Photometric TechniquesFollow the procedure described in the Interfering Factors Test for the Photometric Techniques under Preparatory Testing for the Photometric Techniques.Calculation for the Photometric TechniquesCalculate the endotoxin concentration of each of the replicates of test Solution A using the standard curve generated by positive control series C. The test is not valid unless the following conditions are met: (1) the results of control series C comply with the requirements for validation defined under Verification of Criteria for the Standard Curve under Preparatory Testing for the Photometric Techniques;(2) the endotoxin recovery, calculated from the concentration found in Solution B after subtracting the endotoxin concentration found in Solution A is within 50 to 200%; and (3) the result of negative control series D does not exceed the limit of the blank value required in the description of the LAL Reagent used.Interpretation of Results from the Photometric TechniquesIn photometric assays, the preparation under test complies with the test if the mean endotoxin concentration of the replicates of Solution A, after correction for dilution and concentration, is less than the endotoxin limit for the product.1LAL Reagent reacts with some-glucans in addition to endotoxins. Some preparations thatare treated will not react with-glucans and must be used for samples that contain glucans.2For a validity test of the procedure for inactivating endotoxins, see Dry-HeatSterilization under Sterilization and Sterility Assurance of Compendial Articles1211. Use anLAL Reagent having a sensitivity of not less than 0.15 Endotoxin Unit per mL.3Sterile Water for Injection or other water that shows no reaction with the specific LALReagent with which it is to be used, at the limit of sensitivity of such reagent.4K is 5 USP-EU/kg for any route of administration other than intrathecal (for which K is 0.2USP-EU/kg body weight). For radiopharmaceutical products not administered intrathecally the endotoxin limit is calculated as 175/V, where V is the maximum recommended dose in mL. For intrathecally administered radiopharmaceuticals, the endotoxin limit is obtained by the formula 14/V. For formulations (usually anticancer products) administered on a per square meter of bodysurface, the formula is K/M, where K= 5 EU/kg and M is the (maximum dose/m2/hour × 1.80 m2)/70 Kg.。