戈谢病Ⅱ型1例报道并文献复习
- 格式:pdf
- 大小:1.21 MB
- 文档页数:3

Ⅰ型戈谢病影像表现一例
陈方方;齐先龙
【期刊名称】《影像诊断与介入放射学》
【年(卷),期】2022(31)3
【摘要】戈谢病(Gaucher’s dieasease,GD)是一种罕见的常染色体隐性遗传病,发病率约为十万分之一至五万分之一[1],是一类由葡萄糖脑苷酯酶(glucocerebrosidase,GBA)缺乏引起的溶酶体蓄积病。
可累及骨骼、肝脏、脾脏和中枢神经系统。
目前以骨骼、肝大、脾大受累为主要表现的病例报道居多,以脾大并脾内多发结节灶的病例报道较少见,本文分享一例以脾大并发脾内多发结节灶为主要表现的戈谢病患者的影像表现。
【总页数】3页(P233-235)
【作者】陈方方;齐先龙
【作者单位】山东第一医科大学(山东省医学科学院);济宁市第一人民医院放射科【正文语种】中文
【中图分类】R596;R733.2;R445
【相关文献】
1.梅毒树胶样肿型神经梅毒影像学表现一例
2.天津市一例危重甲型H1N1流感的影像表现
3.右肾上皮样型血管平滑肌脂肪瘤合并囊变和出血影像学表现一例
4.脾脏错构瘤一例病理学分型及影像学表现并文献复习
5.一例枕大孔区不典型毛细胞型星形细胞瘤影像学表现
因版权原因,仅展示原文概要,查看原文内容请购买。

戈谢病疾病研究报告疾病别名:家族性脾性贫血,脑甙病所属部位:全身就诊科室:内分泌科病症体征:急腹症,抽搐,黄疸,吞咽困难,卧床不起疾病介绍:戈谢病是怎么回事?戈谢病是溶酶体贮积病中最常见的一种,为常染色体隐性遗传病,引起不正常的葡萄糖脑苷脂在网状内皮细胞内积聚,法国皮特医生PHILLIPE GAUCHER在1882年首先报道,50年后AGHION报道戈谢病是由于葡糖脑苷脂在肝,脾,骨骼和中枢神经系统的单核-巨噬细胞内蓄积所致,BRADY等在1964年发现葡糖脑苷脂的贮积是由-葡糖苷酶-葡糖脑苷酯酶缺乏所致,为戈谢病的诊断和治疗提供了理论依据症状体征:戈谢病有什么症状?以下就是戈谢病的症状介绍:任何年龄自出生至80岁均可发病,但以少年儿童多发,7岁以下更多。
可分为三型:1、成人型(Ⅰ型)为本病最常见类型,也是脂质贮积病中常见者。
犹太人中多见,但各民族中均有。
在美国估计每年儿童患者不到5000例。
任何年龄均可起病,常以脾脏大就医。
进展可快可慢,进展慢者,脾脏大尤甚,有时有脾梗死或脾破裂而发生急腹症症状。
肝脏呈进行性肿大,但不如脾脏肿大明显。
病程久者,皮肤及黏膜呈茶黄色,常误诊为黄疸,暴露部位如颈、手及小腿最明显,呈棕黄色。
眼球结膜上常有楔形睑裂斑,底在角膜边缘,尖指向内、外眦,初呈黄白色,后变为棕黄色。
肺累及时可影响气体交换而出现症状。
晚期患者四肢可有骨痛,甚而病理性骨折,以股骨下端最常见,也可累及股骨颈及脊柱骨。
有脾功能亢进时可因血小板减少而有出血倾向。
小儿患者身高及体重常受影响。
2、婴儿型(Ⅱ型)患儿自生后即可有肝大、脾大,3~6个月时已很明显,有吸吮、吞咽困难,生长发育落后表现。
神经系统症状突出,颈强直、头后仰、肌张力增高、角弓反张、踺反射亢进,最后变为软瘫,无反应。
脑神经受累时可有内斜,面瘫等症状。
易并发感染。
由于病程短暂,多于婴儿期死亡,因此肝、脾脏肿大不如成人型明显,无皮肤色素沉着,骨骼改变不显著。



戈谢病H型1例报道并文献复习史惠;陈日玲【摘要】目的探讨戈谢病的临床特点及诊断.方法回顾性分析1例经基因学诊断的戈谢病H型患儿的临床特点、骨髓涂片及基因突变类型.结果女孩,7个月,主要表现为肝脾大、角弓反张、精神运动发育落后;骨髓片可见戈谢细胞;分析葡萄糖脑苷脂酶基因,明确突变类型为c.1448T>C(p.Leu483Pro).结论戈谢病H型主要表现为肝脾大,可伴有神经系统受累,葡萄糖脑苷脂酶活性及基因分析对诊断有重要意义.【期刊名称】《中国中西医结合儿科学》【年(卷),期】2019(011)001【总页数】3页(P91-92,0)【关键词】戈谢病;GBA基因;遗传病;儿童【作者】史惠;陈日玲【作者单位】524003 广东湛江,广东医科大学;广东医科大学附属医院儿童医学中心【正文语种】中文【中图分类】R725.5戈谢病又称葡萄糖脑苷脂病,是一种罕见的常染色体隐性遗传病,其遗传方式符合经典的孟德尔理论。
戈谢病是溶酶体贮积症中最常见的一种,由于溶酶体内葡萄糖脑苷酯酶(glucocerebrosidase,GBA)缺乏,导致大量的葡萄糖脑苷脂在肝、脾、骨骼、肺及脑组织的单核-巨噬细胞中贮积[1]。
该病可累及多个系统,临床表现无特异性,极易漏诊或误诊[2]。
广东医科大学附属医院近期接诊1例戈谢病患儿,现回顾性分析该患儿的临床资料,以提高儿科医师对该疾病的认识。
1 临床资料患儿,女,7个月,主因痰鸣3月余,加重伴咳嗽2周,发热2 d入院。
患儿系G2P2,孕39周顺产出生,出生时无窒息抢救史,生后无抽搐病史。
生长发育较同龄儿落后,2个月开始抬头,3个月抬头不稳,5个月会翻身,现7个月,不会独坐,无追人追物反应。
自5月龄开始易呛奶。
父母及哥哥体健,父母非近亲结婚,家族中无类似病史。
入院查体:体温38.3 ℃,脉搏185次/分,呼吸50次/分,体质量6.5 kg,身长65 cm,头围41.5 cm;急性面容,神志清,精神状态一般,颈强直,角弓反张,前囟平软,大小约1 cm×0.5 cm,双侧瞳孔等大等圆,对光反射灵敏,双眼凝视,眼球无水平及垂直运动,牙关紧闭,口唇轻度发绀,咽稍红,呼吸稍促,可见三凹征,以胸骨上窝明显,双肺呼吸音粗,可闻及大量痰鸣音及细湿啰音。

Ⅱ型戈谢病1例并文献复习李尉萌;耿香菊【摘要】Objective To study the clinical and hematology characteristics of gaucher disease.Methods The clinical data of 1 case of gaucher disease of children is analyzed, and the literature review.Results The children asⅡ gaucher disease, has the characteristics of the typical clinical manifestations and hematology.Conclusion This disease is a rare disease, clinical misdiagnosis easily, should pay attention to identify.%目的:探讨戈谢病的临床及血液学特点。
方法对1例戈谢病患儿的临床资料进行分析,并对文献进行复习。
结果此患儿为Ⅱ型戈谢病,具有较典型的临床表现及血液学特征。
结论此病少见,系罕见病,临床易误诊,应注意鉴别。
【期刊名称】《中国继续医学教育》【年(卷),期】2016(008)010【总页数】2页(P51-52)【关键词】戈谢病;罕见病;误诊【作者】李尉萌;耿香菊【作者单位】郑州市儿童医院,河南郑州 450000;郑州市儿童医院,河南郑州450000【正文语种】中文【中图分类】R55戈谢病临床表现为多脏器受累,呈进行性,可危及生命,因其临床表现无特异性,存在漏诊、误诊、误治现象,如能早诊断早治疗,可延缓疾病发展。
本文对其临床特点进行分析,并复习相关文献,以提高认识。
患儿,男,4月。
因发现头后背2月入院。
患儿系足月顺产,无窒息史,黄疸1月消退。
生后不久出现吸气性喉鸣,2月出现头后背。

戈谢病全身骨显像1例解朋;郑晓佐;魏玲格;傅鹏;黄建敏【期刊名称】《中国临床医学影像杂志》【年(卷),期】2019(030)003【总页数】2页(P221-222)【关键词】戈谢病;体层摄影术,发射型计算机,单光子【作者】解朋;郑晓佐;魏玲格;傅鹏;黄建敏【作者单位】河北医科大学第三医院,河北石家庄050051;河北医科大学第三医院,河北石家庄050051;河北医科大学第三医院,河北石家庄050051;河北医科大学第三医院,河北石家庄050051;河北医科大学第三医院,河北石家庄050051【正文语种】中文【中图分类】R596.1;R817.4病例女,13岁,主因“外伤后出现左上肢疼痛2 d”为主诉就诊。
患者于2 d前外伤后出现左上肢疼痛、肿胀及活动受限,未行任何治疗而就诊于我院骨科。
患者一般情况良好,否认家族遗传病史,入院后体格检查示:腹部膨隆,肝脾肋下可及,质地硬。
左上臂轻度肿胀,上臂中段压痛,左肩及左肘关节活动良好。
实验室检查示:WBC:2.99(3.5~9.5)×109/L,红细胞2.33(3.8~5.1)×1012/L,血红蛋白67(115~150)g/L,血小板42(125~350)×109/L,血肌酐50.10(45~84)μmol/L,尿常规未见明显异常。
腹部B超示:肝、脾明显肿大,双肾大小正常,包膜完整,皮质区回声均匀,集合系统无分离。
X线示(图1):左肱骨骨质密度明显减低,骨皮质弥漫性变薄,髓腔扩大,骨小梁模糊,可见多发小虫蚀状骨质破坏区,左肱骨中段可见斜形骨折线,断端无明显错位,考虑为病理性骨折。
遂行石膏固定治疗。
为了解患者全身骨骼情况,而行全身骨显像。
全身骨显像仪器为GE公司产Infinia型SPECT,显像剂采用99mTc-MDP,99Mo-99mTc发生器由中国原子能科学研究院同位素所提供,MDP药盒由江苏原子医学研究所提供,99mTc-MDP标记按说明书进行,放化纯度>95%。

戈谢病戈谢病(Gauchers disease,GD)是溶酶体贮积病中最常见的一种,为常染色体隐性遗传,染色体1q21的GBA基因点突变。
基因异常致β-葡糖苷酶-葡糖脑苷脂酶缺乏,使葡糖脑苷脂在肝、脾、骨骼和中枢神经系统的单核巨噬细胞内蓄积,形成Gaucher cell(戈谢细胞)。
戈谢病在世界各地均有发病,发病率约1/10万-40万,但在某些群体中发病率较高。
根据亚洲地区发病率和临床医生估算,预计中国的戈谢病患者数量不超过 1000 人,但由于戈谢病非常罕见、症状多样,涉及多个科室,医生缺乏认知,已被明确诊断的戈谢病患者人数仅为 300 人,许多患者经历了脾脏切除还没有被正确诊断。
由于β-糖脑苷脂酶缺乏的程度不同,临床表现有较大差异。
1.生长发育落后,甚至倒退。
2.肝脾进行性肿大,尤以脾大更明显,肝功能异常,脾功能亢进,可有淋巴结肿大。
3.骨和关节受累,可见病理性骨折。
4.门脉高压、肺动脉高压。
5.肺受累有咳嗽、呼吸困难和发绀。
6.眼部可见眼球运动失调、斜视、水平注视困难、球结膜对称性棕黄色楔型斑块、基底在角膜边缘、尖端指向眼眦、先见于鼻侧后见于颞侧。
7.皮肤可见鱼鳞病,暴露部位皮肤可见棕黄色斑。
8.中枢神经系统受侵犯可有意识障碍、语言障碍、颈强直、角弓反张、四肢强直、剪刀腿、行走困难、全身肌肉萎缩、牙关紧闭、吞咽困难、喉痉挛、惊厥发作等,脑电图异常。
根据各器官受累的程度,发病的急缓,以及有无神经系统受累,分为3型。
1.Ⅰ型慢性型又称非神经型。
最常见,尤其是犹太人种发病率高,儿童与成人均可发病,以学龄前儿童发病者多,起病缓慢,病程长,无神经系统受累症状。
发病越早,酶活力越低。
通常Ⅰ型患者葡糖脑苷脂酶的活力相当于正常人的12%~45%。
2.Ⅱ型急性型又称神经型。
多在1岁以内发病,最早于生后1~4周出现症状,病情随起病早晚而不同,除Ⅰ型的症状、体征外,神经系统症状明显,多在2岁以前死亡。
此型的葡糖脑苷脂酶活力最低,几乎难以测出。
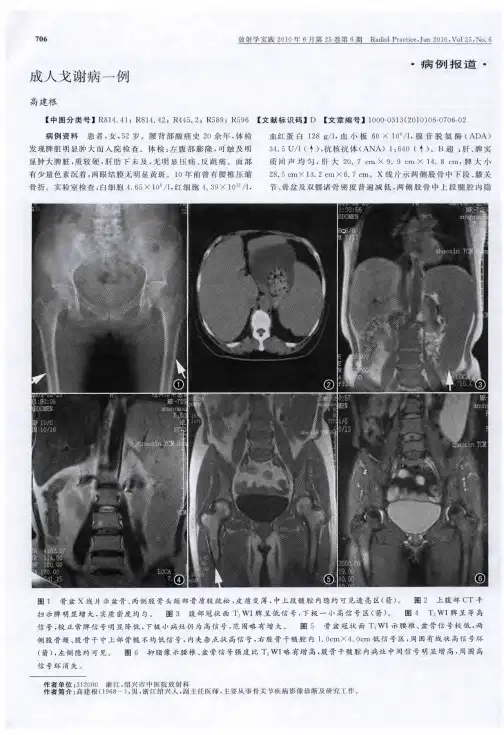

戈谢病1例作者:高晓慧等来源:《今日健康》2015年第03期【关键词】戈谢病1 病例特点患儿,女,1岁0个月。
患儿因“腹部膨隆5月余”入院。
患儿系G1P1,孕40周,剖宫产儿,出生时无窒息。
3月始抬头,8月始独坐,现在12月不会站立。
不会叫爸爸、妈妈。
平时爱哭闹,否认抽搐病史。
患儿父母系近亲结婚。
入院查体:体温:37.2℃,脉搏:126次/分,呼吸:26次/分,头围:44cm,身长:74cm,神志清楚,一般情况尚可,面色苍白,口唇无发绀,浅表淋巴结无肿大,咽部稍充血,双侧胸廓对称,呼吸节律规则,两肺呼吸音粗,未及干湿性罗音。
心律齐,心音力,未闻及心脏杂音。
腹膨隆,按压患儿无哭闹,肝脏肋下5厘米,脾脏至腹股沟,质地中。
脊柱生理弯曲存在,病理反射阴性。
辅助检查资料:网织红细胞计数、异常红细胞形态检查、血细胞分析:白细胞计数 3.79*10^9/L;血小板计数43.00*10^9/L,红细胞计数 3.42*10^12/L,血红蛋白 74.00g/L,尿液分析、粪便常规:未见异常,肝肾功能、心肌酶、电解质:谷丙丙转氨酶78U/L,谷草转氨酶正常,其余正常,凝血功能:未见异常,心电图:窦性心动过速,胸片:双肺纹理增多增粗,心膈未见异常。
全腹CT:1.肝脏稍大,实质密度减低,脾脏增大,代谢性疾病 2.胆囊、双肾、膀胱未见异常。
骨髓细胞学提示:细胞增生明显活跃,粒细胞比例0.43,不同阶段细胞均可见,红细胞比例0.31,可见洋葱皮样细胞约4%,该细胞体积较大,胞质丰富,含大量波纹状、纤维状物质,可见一个或者数个偏心核。
对该细胞进行组织化学染色显示:铁染色:强阳性;糖原染色:强阳性,外送检查葡萄糖脑苷酯酶活性提示:0.5nmol/(g.min),[正常参考范围:2.63—25.6 nmol/(g.min)]。
提示葡萄糖脑苷酯酶活性明显下降。
综合上述资料分析,该患儿戈谢病诊断明确。
2 讨论戈谢病(Gaucher disease,GD)属于先天性常染色体隐性遗传性疾病[1-2],为溶酶体贮积病中较常见的一种。

可治性罕见病—戈谢病一、疾病概述戈谢病(Gaucher disease)是一种罕见的常染色体隐性遗传代谢类疾病,主要是由于编码葡萄糖脑苷脂酶(acid p - Glucocerebrosidase,GBA)的基因突变引起的常染色体隐性遗传病。
由于基因突变,造成体内该酶活性下降,从而导致其底物葡萄糖脑苷脂不能被正常降解而在溶酶体中贮积,在多种组织中形成典型的溶酶体贮积细胞即戈谢细胞,临床表现为多脏器受累并呈进行性加重,甚至危及生命。
1882年,由法国医生Gaucher首先报道而得名。
该病发病率在不同种族间有很大差异,东欧犹太阿什可纳济人根据杂合子频率预测该病发病率高达1/450[1.2]。
普通人群该病发病率在11100 000~1/40 000[3-5]。
二、临床特征根据神经系统是否受累,将戈谢病主要分为非神经病变型(I型)及神经病变型(Ⅱ型及Ⅱ型)[6]。
其他少见亚型(围产期致死型、心血管型[7]等)也有报道。
1、Ⅱ型(非神经病变型)最常见,无原发性中枢神经系统受累表现。
各年龄段均可发病,主要为肝脾大,尤以脾大显著,可伴脾功能亢进。
血液学主要表现为血小板计数减少及贫血,甚至白细胞计数减少。
患者可有疲乏无力、皮肤及牙龈出血,支性可表现出月经量增多。
患者可有急性或慢性骨痛,严重者出现骨危象(严重骨痛急性发作,伴发热及白细胞计数增高、ESR加快)。
X线检查表现为股骨远端的烧瓶样畸形、骨质减少、骨质疏松,重者出现骨的局部溶解、骨梗死、病理性骨折、关节受损等。
骨骼病变可影响日常活动,并可致残。
部分患者可有肺部受累,主要表现为间质性肺病、肺实变、肺动脉高压等。
2、Ⅱ型(急性神经病变)婴儿期发病,有迅速进展的癫痫发作、角弓反张等急性神经系统受损表现,精神运动发育落后,一般于2~4岁前死亡。
3、Ⅱ型(亚急性神经病变)也称慢性神经病变,早期表现与Ⅱ型相似,逐渐出现神经系统表现,病情进展缓慢,寿命可较长。
患者常有动眼神经受侵、眼球运动障碍[8]。
戈谢病12例临床分析
王秀敏;梁黎
【期刊名称】《浙江预防医学》
【年(卷),期】2005(017)004
【摘要】戈谢病(Gaucher’s disease)是一种常染色体隐性遗传病,是由于溶酶
体8-葡萄糖脑苷酶缺陷所引起的类脂质贮积病,虽发病率低(1/10万-1/40万),但对儿童全身影响较大。
现对12例儿童戈谢病进行临床报告如下。
【总页数】2页(P43,50)
【作者】王秀敏;梁黎
【作者单位】浙江大学医学院附属儿童医院,浙江,杭州,310003;浙江大学医学院附
属儿童医院,浙江,杭州,310003
【正文语种】中文
【中图分类】R725.9
【相关文献】
1.戈谢病3例临床分析及文献复习 [J], 唐家彦;薛湘萍;刘玉玲;付四毛
2.125I粒子植入治疗前列腺癌122例临床分析 [J], 张春霆;党瑞锋;徐勇;刘冉录;张志宏;杨阔;马宝杰;任海林;乔宝民
3.128层螺旋CT诊断120例气管憩室的临床分析 [J], 庞旭曼;李要京;李强;于永波;周荣林
4.2008年至2012年某地区手足口病1223例临床分析 [J], 柳红花
5.戈谢病5例临床分析 [J], 余思邈; 王睿林; 朱云; 王立福; 张宁; 孙永强; 景婧; 桑秀秀; 王丽苹; 许文涛
因版权原因,仅展示原文概要,查看原文内容请购买。
Ⅰ型戈谢病并发病理性骨折1例
叶祥
【期刊名称】《医学影像学杂志》
【年(卷),期】2012(022)007
【总页数】2页(P1177,1181)
【作者】叶祥
【作者单位】江苏省南京下关中医医院放射科江苏南京 210011
【正文语种】中文
【中图分类】R589.2;R445
【相关文献】
1.戈谢病引起的胫腓骨病理性骨折1例 [J], 黄占国;王晓桐
2.以股骨中段病理性骨折为首发表现的成人戈谢病1例 [J], 王志敏;张惠箴;杨庆城;章振林
3.成人Ⅰ型戈谢病合并股骨干病理性骨折、股骨头坏死1例 [J], 顾军;谢林;贾晋辉;王庚启
4.四肢骨巨细胞瘤伴病理性骨折的外科治疗及并发症 [J], 文立;乔军;孟凡青;王冬梅;陈骏;陈亭亭;王雪迪;王守丰
5.多发性骨髓瘤IgA Ⅲ期λ型并发寰枢椎病理性骨折1例报告及文献回顾 [J], 连笑宇;马浩浩;邵佳;高坤;高延征
因版权原因,仅展示原文概要,查看原文内容请购买。