冻干粉针剂工艺
- 格式:docx
- 大小:36.57 KB
- 文档页数:16

关于冻⼲粉针⼯艺流程
关于冻⼲粉针⼯艺流程,主要⼯艺流程:分装好药液的安瓿或西林瓶→预冻→升华⼲燥→再⼲燥预冻:预冻是恒压降温的过程。
升华⼲燥:冷冻⼲燥的主要过程,升华的条件:1、保证冰不熔化2、冰周围的⽔蒸⽓压低于饱和蒸汽压(有两种⽅法:⼀次升华法和反复冷冻升华法)
再⼲燥:升华完成后,物料内留下许多空⽳,单物料的机制内还留有残余的未冻结⽔分10%左右,温度继续升⾼⾄0℃或室温,并保持⼀段时间,可使升华的⽔蒸⽓或残留⽔分被抽尽。
在⼲燥可保证冻⼲制品含⽔量注意事项:
1、含⽔量偏⾼
装⼊容器的药液过厚,升华⼲燥过剩中供热不⾜,冷凝器温度偏⾼或真空度不够,均可能导致含⽔量偏⾼。
可采⽤旋转冷冻机及其他相应的⽅法解决。
2、喷瓶
如果供热太快,受热不均匀或预冻不完全,则易在升华过程中使制品部分液化,在真空减压条件下产⽣喷瓶。
为防⽌喷瓶,必须控制预冻温度在共熔点以下10~20℃,同时加热升华,温度不宜超过共熔点。
3、产品外形不饱满或萎缩
⼀些黏稠的药液由于结构过于致密,在冻⼲过程中内部⽔分逸出不完全,冻⼲结束后,制品会因潮解⽽萎缩。
遇到这种情况通常可在处⽅中加⼊适量的⽢露醇、氯化钠等填充剂,并采取反复预冻法,以改善制品的通⽓性,产品外观即可得到改善。

冻干粉针工艺冻干粉针是将药品的无菌溶液快速冻结到零下40度左右,然后在真空条件下,慢慢加热,使溶液的水分升华,同时保持冻结状态无菌粉注射剂。
其常见的生产工艺是:先将药物按水针剂标准制成药液,在百级状态下经过滤、超滤除去杂质和细菌,无菌分装入瓶半加塞,装入冻干机迅速冷冻至零下40度左右使药液成固态,在此状态下抽极限真空(<0.009Pa)将水分升华排出,药物有序结晶干燥成膨松晶体,升华完成后在真空状态下自动加塞,则无菌、真空、含水量<0.01%的晶态冻干粉针就生产成了。
因而在贮藏上一般的冻干粉针无需冷藏,常温保存即可。
冻干粉针与一般粉针和水针剂比稳定性好:因为冻干粉针是在真中试冻干机Pilot2-4L博医康空状态下制成和保存的,真空状态几乎无氧气,药品不会被氧化;冻干粉针含水量极低(<0.01%),不会被水解;冻干粉针制作过程严格无菌(达百级标准),不会被污染,所以2冻干粉针稳定性好。
冻干粉针也比一般粉针起效快,生物利用度高。
这是因为冻干粉针特殊的生产工艺使药品均匀分散在由壳聚糖等冻干形成的冰架内,此结构扩大了药物的表面积,使得药物在单位时间内能接触到更多病菌,从而具有速效功能。
壳聚糖形成的特殊冰架结构可使药品呈纳米状态存在于一个个微囊中,随血液循环到达病灶部位释放出来,提高了药物的生物利用度。
冻干粉针的高科技体现在:冻干粉针除真空、无菌、含水量极低、制作过程速冻.升华等科技含量较高外,还有其以下高科技含量:(1)可使药物微囊化或形成脂质体:通过筛选冻干载体、调整冰架形状和微孔大小,可使药物微囊化或形成脂质体,通过筛选不同的微囊和脂质体,可使药物达到速效、靶向或控制释放作用。
(2)通过调整载体和冻干工艺,可使两种药物在特定的环境条件下复合成复盐,后者可使两种药物同时抵达病灶,发挥协同作用,大大提高了抗菌药物的杀菌效能。
且复盐使药物稳定性增强。
之所以把药物制成冻干粉针原因在于:(1)有些药物稳定性差,或见水很容易分解,不能作成普通粉针或水针,只有作成冻干粉针才能很好发挥药效。

冻干粉针剂工艺流程Freeze-drying Powder Injection Process。
Freeze-drying technology is widely used in the pharmaceutical industry to produce powder injection drugs. The process involves several steps, including pre-treatment, freezing, primary drying, secondary drying, and packaging. Each step is crucial to ensure the quality and stability of the final product.Pre-treatment:Before the freeze-drying process, the drug solution needs to be pre-treated to remove impurities and adjust the pH value. The solution is filtered through a sterile filter to remove any bacteria or particles. Then, the pH value is adjusted to ensure the drug's stability during the freeze-drying process.Freezing:After pre-treatment, the drug solution is filled into vials or ampoules and then quickly frozen at a low temperature. The freezing process is critical to ensure the formation of a uniform ice crystal structure. The uniform ice crystal structure can prevent damage to the drug molecules during the drying process.Primary Drying:The primary drying process involves sublimation, where the frozen water in the drug solution changes from a solid to a gas without going through the liquid phase. The process is conducted under a low-pressure environment and a low-temperature condition. The primary drying process can take several hours to several days, depending on the drug's characteristics and the equipment used.Secondary Drying:After the primary drying process, the drug productstill contains some residual moisture. The secondary dryingprocess is designed to remove the remaining moisture and improve the drug's stability. The temperature and pressure conditions are adjusted to ensure the complete removal of moisture without damaging the drug molecules. The secondary drying process can take several hours to several days, depending on the drug's characteristics and the equipment used.Packaging:After the secondary drying process, the drug product is sealed in vials or ampoules under sterile conditions. The packaging process is critical to ensure the drug's quality and stability during storage and transportation.In conclusion, the freeze-drying powder injection process is a complex and delicate process that requires strict control of temperature, pressure, and time. The process can produce high-quality and stable powderinjection drugs with a long shelf life. The quality and stability of the final product depend on each step of the process, from pre-treatment to packaging. Therefore, it isessential to follow the standard operating procedures and conduct rigorous quality control throughout the process.。

一、冷冻干燥过程研究真空冷冻干燥是先将制品冻结到共晶点温度以下,使水分变成固态的冰,然后在适当的温度和真将水蒸气冷凝,从而获得干燥制品的水汽凝结器)空度下,使冰升华为水蒸气。
再用真空系统的冷凝器(、二次干燥(解吸干燥)和密封保技术。
该过程主要可分为:制品准备、预冻、一次干燥(升华干燥)存五个步骤。
1 产品预冻制品的玻璃化1.1玻璃化的作用。
近年来,人们已经逐渐地认识到,凡是成功的低温保存,细胞的水均以玻璃态的分子玻璃化是指物质以非晶态形式存在的一种状态,其粘度极大,形式被固化,在胞不出现晶态的冰。
故在这种结构中分子运动和分子变性反应非的能动性几乎为零,由于这种非晶体结构的扩散系数很低,常微弱,不利的化学反应能够被抑制,从而提高被保存物质的稳定性。
玻璃化的获得。
在产品预冻时,只要降温速率足够快,且达到足够低的温度,大部分材料都能从指“足够快”的意思是在降温过程中迅速通过结晶区而不发生晶化,“足够低”液体过冷到玻璃态固体。
Tg 以下。
的是必须把温度降到玻璃化转变温度第一步是以一般速对于具有一定初始浓度的细菌制品,其预冻过程一般通过“两步法”来完成。
第胞溶液的浓度逐渐提高;,让细胞外的溶液中产生冰,率进行降温细胞的水分通过细胞膜渗向胞外,,以实现胞溶液的玻璃化。
此法又称“部分玻璃化法”。
二步是以较高速率进行降温点后将开BA当初始浓度为A的溶液(点)从室温开始冷却时,随着温度的下降,溶液过冷到冰晶周围溶液将沿着平衡的熔融线不断析出冰晶,始析出冰,结晶潜热的释放又使溶液局部温度升高。
(D的交点(Ta)与玻璃化转变曲线(Tg)剩余的未冻溶液随温度下降,浓度不断升高,一直下降到熔融线,此时的溶液达到最大冻结浓缩状,浓度较高,)时,溶液中剩余的水分将不再结晶(称为不可冻水点) 以非晶态的形式包围在冰晶周围,形成镶嵌着冰晶的玻璃体。
降温速率与预冻温度1.2其速度可控制在每分钟降形状和成品最初晶格及其微孔的特性,预冻速度决定了制品体积大小、1℃左右。

1概述冻干粉针剂是将药物的灭菌水溶液无菌灌装后,进行冷冻干燥而制成的注射用粉末。
其制剂工艺特点:①对制剂的无菌要求非常高;②药物中的溶剂是在低温(-50℃~30℃)低压(6~10Pa)从固态不经过液态而直接升华为气态而除去的。
由于冻干粉针剂含水量低,干燥又在低温真空中进行,故不易被氧化,有利于药品的长期贮存,特别适用于性质不稳定,不耐热等药物的生产制备。
但是该生产工艺对无菌条件要求非常高,需要在硬件设施和生产管理等各个方面严格控制。
最大限度地减少各种污染可能,确保产品的无菌要求。
通过多年的工程设计经验积累和对GMP的不断学习,针对冻干粉针车间的工艺设计,笔者有以下几点感受提出来与大家共同商榷。
2工艺布局厂房是药品生产的根本条件,在GMP认证检查中占据重要的位置。
WHO的GMP对厂房要求的原则是:厂房选址、设计、施工、改造和保养适合生产操作。
其布局及设计必须以降低差错的危险性和能有效地清洁和保养为目标,为的是避免交又污染。
避免对产品质量有任何不良影响。
《药品生产质量管理规范(1998年修订)》(以下简称GMP)第九条明确指出:“厂房应按生产工艺流程及所要求的空气洁净级别进行合理布局”。
这就要求厂房工艺布局按生产过程和操作程序,做到物顺其流,人行其畅。
既物料按生产流程顺序,以最短的路线传递,避免往返交叉。
工作人员上岗路线尽量短,不穿岗,避免迂回曲折。
最大限度地减少差错和交叉污染。
工艺布局不当不仅会导致操作不便,人流物流混乱,造成差错、污染,也不利于设备的安装、清洗、维护、检修,而巨影响净化空调的气流组织,增加能源消耗和建设成本。
3设备选型与布置GMP第四章对设备的设计、选型、安装作出了原则性规定,为了保证药品质量,更好地执行GMP,在设备选型方面应注意以下几点:(1)所选设备要符合药品生产和工艺要求。
结构简单、表面光洁,便于操作和维护保养。
(2)与药品直接接触的设备内表面应选用不与药品反应、不释出微粒、不吸附物料的材质。
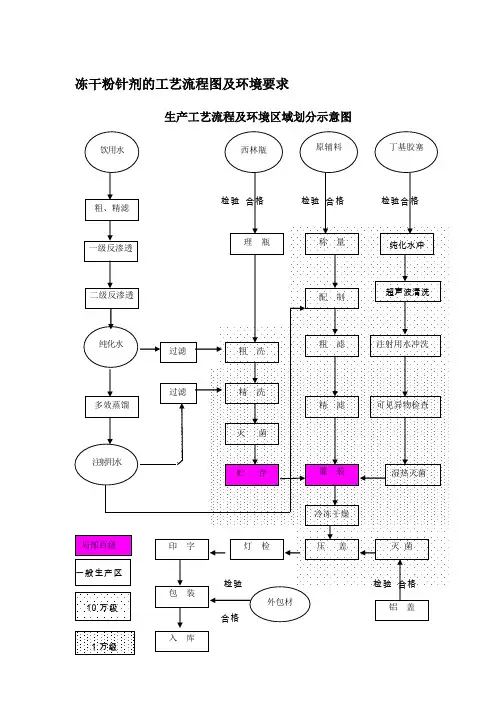
冻干粉针剂的工艺流程图及环境要求生产工艺流程及环境区域划分示意图入 库冻干粉针剂认证检查的要点冻干粉针剂指在低温真空的环境下先将配制好的药液预冻然后升华干燥成粉末状态的一种制剂。
根据药品新版GMP原则要求,结合冻干粉针剂生产影响药品质量的关键环节,提出冻干粉针剂GMP检查要点,旨在督查药品生产企业质量保证体系和实际运行状态。
检查组须对如下内容按照药品GMP要求,逐一核实。
1、机构与人员a)主管生产和质量管理的企业负责人、生产管理和质量管理部门负责人均应具有医药或相关专业大专以上学历,并具有药品生产和质量管理经验,并履行其职责。
b)企业负责人和各级管理人员应定期接受药品管理法律法规培训。
c)质量检验、生产、维修保养、清洁人员应定期进行卫生和微生物学基础知识、洁净作业等方面的培训和考核,并具有实际操作技能。
2、厂房设施的管理要点及检查重点a)洁净区:我国《药品生产质量管理规范》对最终灭菌的无菌药品生产厂房洁净度级别的要求是:浓配或采用密闭系统的稀配应在100000级洁净区内进行;稀配、滤过、灌封、直接接触药品的包装材料的最终处理等操作应在10000级洁净区内进行;物料、中间品应经过物流缓冲间或传递柜进、出洁净区。
称量配料间如产尘应与洁净走廊呈相对负压,必要时设捕尘设施。
中国药典规定:微生物限度检查、无菌检查应在100级或10000级背景下的局部100级区内进行,并与生产区分开。
微生物限度检查与无菌检查用的实验室和空气净化系统最好彼此分开,以尽可能减少对无菌检查的干扰。
b)空气净化系统应能确保洁净区的洁净度级别、温湿度、压差等符合生产工艺要求并经过验证。
初效、中效过滤器应明确清洗/更换周期,高效过滤器应定期检测其完整性,如有泄漏或阻塞应及时更换。
空气净化系统应每天24小时运行,停用后再次运行应进行清洁、消毒并经过再验证,符合要求的方可开始生产。
c)与产品直接接触的压缩空气、氮气、二氧化碳等辅助设施这些气体因与产品直接接触,不得对产品带来污染。

皖西学院化学与生命科学系(制药设备与工程设计)课程设计班级姓名学号指导教师二○一○年六月二十一日制药设备与工程设计课程设计任务书皖西学院化学与生命科学系课程设计说明书题目:课程:系(部):专业:班级:学生姓名:学号:指导教师:完成日期:目录1.工艺流程图及环境区域划分2.生产处方及其依据3.生产工艺及操作程序4.设备一览表及主要设备生产能力5.技经指标与消耗定额6.劳动定员7.技术安全、劳动保护和工艺卫生8.物料平衡1.粉针剂工艺流程图及环境区域划分图例:100 000级区 10 000级区 * 局部百级2.产品介绍注射用青霉素钠,英文名:Benzylpenicillin Sodium for Injection 。
主要成份为青霉素钠,其化学名为(2S,5R,6R)-3,3-二甲基-6-(2-苯乙酰氨基)-7-氧代-4-硫杂-1-氮杂双环[3.2.0]庚烷-2-甲酸钠盐。
性状本品为白色结晶性粉末。
药理作用为β-内酰胺类抗生素,系通过干扰细菌细胞壁粘肽的合成, 于细菌繁殖期起杀菌作用。
主要作用于革兰氏阳性菌、革兰氏阴性球菌、嗜血杆菌属及螺旋体、放线菌等。
青霉素内服后在胃酸中大部分灭活,一般剂量达不到有效血浓度。
其钠盐肌肉注射后吸收迅速,约0.5h达血药浓度,对多数敏感菌的有效血浓度可维持6~8h。
青霉素在血中约有50%以上(马为52~54%)与血浆蛋白结合。
在血中未结合的和游离的青霉素能通过被动扩散分布于组织、体液中,但不易透入眼、骨组织、脑脊液和浓肿腔中,易透入有炎症的组织。
青霉素约50~75%自肾脏排出,尿中浓度也很高。
丙磺舒、磺胺药、阿司匹林等可提高其血药浓度,延长其半衰期。
本品为抗生素类药。
主用于革兰氏阳性菌感染,亦可用于放线菌及钩端螺旋体等的感染。
2.1产品规格注射青霉素钠(0.3、0.5g、0.6g、)2.2产品剂型、规格和批量3.生产工艺及操作程序3.1生产场所和设备3.1.1主要操作间的位置和要求3.1.2主工生产设备型号和编号3.2关键生产设备操作规程和清洁规程3.3生产步骤3.3.1原料的准备工作:在无菌分装操作前一天,xxxx无菌原料经物料员凭批生产指令从仓库领取后,在缓冲间擦拭干净,由物料员和QA检查员依据领料核料单审核原料名称,规格,批号、重量,是否有检验合格证等,审核合格后,由车间生产人员用消毒液揩擦桶外壁后,放到物料传递间原料经净化后传入万级B区,第二天方可经传递窗紫外灯照射30分钟后传入万级A区。

冻干粉针剂工艺流程
《冻干粉针剂工艺流程》
冻干粉针剂是一种常见的制剂形式,它可以延长药物的保存时间并方便患者使用。
其制备工艺流程主要包括以下几个步骤:
1. 原料准备:首先需要准备药物原料,以及辅料如稳定剂、缓冲剂等。
这些原料需要经过严格的质量检验和筛选,确保其符合制剂要求。
2. 溶液调制:将药物原料和辅料按照一定的配方和比例加入溶剂中,并充分溶解,形成均匀的混合溶液。
3. 过滤消毒:对溶液进行过滤,去除其中的杂质和微生物,确保制剂的纯净度和无菌性。
4. 注射制剂:将过滤后的溶液充入注射器中,并根据计量要求进行分装,确保每支注射器中的药物量准确无误。
5. 冷冻干燥:将注射器中的溶液置于冷冻干燥设备中,通过冷凝、真空等技术将其中的水分逐渐去除,直至形成干燥的粉末。
6. 包装贮藏:将冻干粉装入密封的药品包装中,并在干燥、低温条件下储存,以确保其质量和稳定性。
通过以上工艺流程,冻干粉针剂可以在保持药物活性的同时,
保持较长的有效期限,方便患者使用。
同时,严格的生产工艺和质量控制,也能够保证冻干粉针剂的稳定性和安全性。

冷冻干燥制备冻干粉针剂的工艺流程下载提示:该文档是本店铺精心编制而成的,希望大家下载后,能够帮助大家解决实际问题。
文档下载后可定制修改,请根据实际需要进行调整和使用,谢谢!本店铺为大家提供各种类型的实用资料,如教育随笔、日记赏析、句子摘抄、古诗大全、经典美文、话题作文、工作总结、词语解析、文案摘录、其他资料等等,想了解不同资料格式和写法,敬请关注!Download tips: This document is carefully compiled by this editor. I hope that after you download it, it can help you solve practical problems. The document can be customized and modified after downloading, please adjust and use it according to actual needs, thank you! In addition, this shop provides you with various types of practical materials, such as educational essays, diary appreciation, sentence excerpts, ancient poems, classic articles, topic composition, work summary, word parsing, copy excerpts, other materials and so on, want to know different data formats and writing methods, please pay attention!一、概述冻干粉针剂是一种常见的固体制剂形式,其制备工艺过程主要包括冷冻和干燥两个步骤。

冻干粉针剂工艺流程冻干粉针剂是一种常见的药物剂型,其工艺流程包括原料准备、混合、充填、冻干、密封等多个环节。
下面将详细介绍冻干粉针剂的工艺流程。
首先是原料准备。
冻干粉针剂的原料包括药物原料、辅料和溶剂。
药物原料应当符合药典规定的质量标准,辅料应当具有良好的稳定性和流动性,溶剂应当纯净无菌。
在原料准备环节,需要对原料进行严格的检验和筛选,确保其符合生产要求。
接下来是混合环节。
在混合环节中,需要将药物原料、辅料和溶剂按照一定的配方比例加入混合容器中,并进行搅拌混合,直至混合均匀。
混合的均匀性对于冻干粉针剂的质量至关重要,因此在混合环节中需要严格控制搅拌时间和速度。
然后是充填环节。
在充填环节中,需要将混合好的药物溶液充填到预先准备好的注射器中。
充填时需要注意避免气泡的产生,保证充填的药物溶液质量完整,这对于后续的冻干环节非常重要。
接着是冻干环节。
在冻干环节中,需要将充填好药物溶液的注射器放置在冻干机内,进行冻干处理。
冻干的目的是将溶剂从药物溶液中去除,使药物呈现出干粉状态。
冻干的过程需要严格控制温度和压力,确保冻干的质量。
最后是密封环节。
在密封环节中,需要将冻干后的药物溶液密封在注射器中,保证其不受外界环境的影响。
密封环节需要使用高效的密封设备,确保密封的牢固性和密封后的产品质量。
以上就是冻干粉针剂的工艺流程,每个环节都需要严格控制,确保产品的质量和安全性。
冻干粉针剂作为一种常见的药物剂型,在医疗领域有着广泛的应用,其工艺流程的完善和优化对于产品的质量和效果都有着重要的影响。

冻干粉针剂冻干工艺及质量技术探讨摘要:冻干粉针剂是近些年来药品制剂中的创新剂型,对于蛋白质、微生物类等易发生变性、易丢失生物活性的药品尤为适用,具有不易氧化、不易水解、稳定性更高、生物利用度高、起效快和容易配伍等优点。
本文对冻干粉针剂的冻干工艺、质量技术两方面进行探讨。
关键词:冻干粉;针剂;冻干工艺;质量技术前言:冻干粉针剂因为其优点在临床中的需求不断扩大,冻干工艺因此也得到了飞速的发展,但是在实际的冻干过程中仍存在一些问题。
随着社会的发展,对产品需求的提高、人们环保意识的提高和能源环境问题的重视等原因,对冻干粉针剂的工艺及质量技术不断提出了新的要求,需要不断的探讨和发展。
一、冻干粉针剂冻干工艺冻干粉针剂是在无菌环境下使用真空冷冻干燥机使得药液经过冷冻、水分升华等过程,最终得到保留活性的无菌粉注射剂。
这一过程主要可以分为制品准备阶段、产品预冻阶段、药物升华干燥阶段、二次解析干燥阶段以及密封保存等五个阶段。
(一)制品准备阶段制品的准备阶段是影响冻干药物质量的重要环节,严格的药物制备环节可以降低药物冻干过程中的干扰因素。
制品准备首先按照质量标准将药物制成水针剂药液,并以百级标准进行过滤去除杂质和细菌,无菌条件下分装至西林瓶、安剖瓶等容器内,再将已灌装好的中间产品放入真空冷冻干燥机内,完成制品准备阶段的内容。
(二)产品预冻阶段在做好药物制备工作后,可以进入到产品预冻阶段,此阶段的目的是使得产品定型,是后续真空升华工序的基础,是影响药物冻干质量的重要环节,不良的预冻会影响药品的质量、降低部分药品的活性以及增加生产时间。
不同药物溶液的冷冻速度不同,需根据制剂本身的特性决定,如一般疫苗类药品制成冻干粉的过程中,通常采取快速降温的方式较为有利,而对于蛋白多肽类药物冻干粉的制作,通常采取慢速冻结方式。
(三)药物升华干燥药物的升华干燥又称为一次干燥,在真空低温环境下通过对药品加热,使得药品所含的水分直接升华为水蒸气去除大部分水分。
粉针剂冷冻干燥工艺及质量技术探讨冻干粉针剂是药品研发中的一种新的类型,冻干粉针剂的制备需要在低温下展开,非常适用于很多热敏性物质。
微生物、蛋白质等在冻干制备过程中生物活性不会丧失改变,正是因为这些特点,近年来在临床上有着非常广泛的应用,具有非常广阔的发展前景。
冻干粉针剂在生产过程中对于冻干工艺有着十分严格的要求,必须要提高其质量技术应用规范性和有效性,使冻干粉针剂生产质量得到保证,本文就此展开了研究分析。
1 冻干粉针剂1.1 冻干粉针剂发展现状早在二十世纪三十年代,国内微生物学家开始对盐水预冻展开研究,对蒸发器进行抽真空处理,之后采取吸水剂方式制作冻干菌毒种,并将其保存。
之后在二十世纪五十年代,国内很多企业开始进行冻干疫苗的批量生产,主要应用在动物方面。
在六七十年代,有部分企业开始将该项技术应用在冻干疫苗以及血浆生产中,从此冷冻干燥技术真正意义上开始应用在我国制药以及食品行业。
1.2 冻干粉针剂定义冻干粉针剂是指对无菌药品溶液迅速降温处理,置于真空加热状态,通过液体的升华特性将其中的水分去除,生产出有着非常好冻结状态的无菌粉注射剂,满足临床实际使用需求。
1.3 冻干粉针剂特点与常见粉剂以及水剂相比,冻干粉剂有着非常好的稳定性,含水量最低可以控制在0.01%以下,不会被水解。
在进行冻干粉针剂的生产时需要严格按照GMP规范中的无菌要求进行,避免被污染。
冻干粉针药品在实际应用中往往会较为均匀地分布在冻干冰架内,从结构角度而言会一定程度上增大药物表面积,起效速度快于一般的粉针,生物利用度能够有显著提高。
另外,冻干粉针剂的科技含量相对较高,能够通过一定的技术手段使药物形成脂质体或者微囊化,作用于人体迅速达到靶器官,还能够起到控制释放效果。
生产过程中还会结合实际需要对药物生产工艺以及冻干载体进行适当的调整,保证两种药物能够在特殊条件下复合,发挥协同作用,提高药物治疗效果。
2 冻干粉针剂工艺控制冻干粉针剂类药品按照报批的生产工艺生产,根据报批处方配制后形成无菌药液,按规格进行分装/灌装,之后放入冷干设备进行冷冻干燥,冷干设备以真空冷冻干燥机为主,即冻干机。
冻干粉针生产工艺流程英文回答:Lyophilization of Injectable Powders: A Comprehensive Process Walkthrough.Lyophilization, also known as freeze-drying, is a specialized process widely employed in the pharmaceutical industry to preserve and enhance the stability ofinjectable powders. It involves the sublimation of water from a frozen product under a vacuum environment, resulting in the formation of a dry and shelf-stable powder. This process offers several advantages, including improved product stability, prolonged shelf life, ease of reconstitution, and enhanced bioavailability.Process Overview:The lyophilization process typically consists of several key stages:1. Formulation Development:The initial stage involves formulating the product to be lyophilized. This includes determining the optimal composition of the drug substance, excipients (e.g., bulking agents, cryoprotectants), and solvents.2. Freezing:The formulated product is then filled into vials or other suitable containers and subjected to a controlled freezing cycle. The freezing rate and temperature are carefully regulated to ensure the formation of small, uniform ice crystals throughout the product.3. Primary Drying (Sublimation):The frozen product is placed in a specialized lyophilizer, where it is exposed to a vacuum. The vacuum environment lowers the atmospheric pressure, causing the ice crystals to sublime directly into water vapor withoutpassing through the liquid phase. This sublimation process effectively removes the majority of the water content from the product.4. Secondary Drying (Desorption):Once the primary drying is complete, the product still contains a small amount of residual water bound to the drug substance and excipients. Secondary drying involves raising the temperature of the product while maintaining the vacuum. This step removes the remaining water by desorption, ensuring the formation of a dry and stable powder.5. Reconstitution:The lyophilized powder is typically intended for reconstitution with a suitable solvent (e.g., water for injection) before administration. The reconstitutionprocess involves adding the solvent to the powder, which rapidly dissolves to form a solution or suspension.Benefits of Lyophilization:Lyophilization offers several significant benefits for the production of injectable powders:Enhanced Stability: Lyophilized powders exhibit improved stability compared to liquid formulations, making them suitable for prolonged storage at room temperature or above.Extended Shelf Life: The removal of water effectively inhibits microbial growth and chemical degradation, extending the shelf life of the product.Ease of Reconstitution: Lyophilized powders are easy to reconstitute, requiring minimal preparation and providing a convenient format for healthcare professionals and patients.Improved Bioavailability: In certain cases, lyophilization can enhance the bioavailability of the drug substance by improving its solubility and absorption.Applications:Lyophilization is extensively used in the production of various injectable powders, including:Antibacterial and antiviral agents.Antibiotics.Vaccines.Enzymes.Hormones.Conclusion:Lyophilization is a highly effective process for the production of stable and shelf-stable injectable powders.Its ability to preserve the integrity of the drug substance, extend shelf life, and enhance bioavailability makes it a valuable tool in the pharmaceutical industry. The precisecontrol of freezing, sublimation, and desorption processes ensures the production of high-quality powders with optimal stability and efficacy.中文回答:冻干粉针生产工艺流程。
冻干粉针剂生产工艺流程
冻干粉针剂生产工艺流程一般包括以下步骤:
1. 原料准备
准备所需原材料,包括药物活性成分、辅料、溶剂等。
2. 制备溶液
将药物和辅料溶解在适当的溶剂中,形成混合溶液。
3. 过滤
将混合溶液经过滤器过滤,去除杂质、微生物或不溶性物质。
4. 均匀冻结
将过滤后的溶液倒入冷却的托盘中,以均匀的方式冻结。
冻结速率和方向需加以控制,以确保冻结温度和冻结时间相同。
5. 预处理
在脱水前,进行预处理,如抑制氧化、杀菌等。
6. 脱水
将托盘放入真空脱水装置中进行脱水,以去除冰晶。
脱水过程可以通过增加托盘温度、加大真空度等方式实现。
7. 冷降温
将干燥的冻干物进行降温,以防止水分重新结晶。
8. 自动装填
将冻干物装填进针剂容器中,如玻璃瓶、注射器等。
9. 质量控制
对成品进行质量控制,包括外观、悬浮液的稳定性、pH值、活性成分含量等方面。
10. 包装
将成品进行包装,通常采用塑料薄膜袋或铝箔袋密封包装,以防止水分、氧气或污染物的进入。
冻干粉针剂生产工艺验证方案冻干粉针剂是一种常见的药品剂型,其生产工艺涉及多个环节,需要严格验证方案确保产品质量和安全性。
本文将从生产流程、验证方案和实验设计等角度,探讨冻干粉针剂生产工艺验证方案的编制及实施。
一、生产流程冻干粉针剂的生产流程一般由以下步骤组成:原材料检验、制剂、灌装、冷冻干燥、包装。
其中冷冻干燥是制备过程中最重要的环节。
冷冻干燥过程是指将含有大量水分的制品冷冻后,在低压条件下,通过升温、升压等方式使冻结的水分逸出,从而得到干燥的产品。
冷冻干燥的过程不仅会影响产品质量和产量,还涉及到能源消耗、生产成本和工艺设备的选择等方面。
二、验证方案制定有效的冻干粉针剂生产工艺验证方案,能够保证产品的质量和安全性。
验证方案需要涵盖产品制造过程中的各个环节和关键点,并设立一套严格的实验设计和实验方法,进行各项验证实验和数据分析。
1.验证目标:验证方案需要明确验证的目标,包括制备工艺的可控性、生产设备的稳定性和生产过程的可重复性等方面。
2.工艺要求:工艺要求是冻干粉针剂生产过程中必须满足的条件和指标,如产品水分含量、制剂时间、注射器尺寸等。
3.实验设计:实验设计是验证方案中最为重要的环节之一。
合理的实验设计可以减少实验成本和时间,提高实验效率。
实验设计包括试验方法、操作程序、研究某个变量对产品质量的影响等方面。
4.数据分析:数据分析是验证方案中的最后一步,对实验结果进行分析和判断。
数据分析需要综合考虑试验方案、原始数据和实验方法等因素。
三、实验设计冻干粉针剂的生产工艺验证实验,需要有一套合理的实验设计来指导和管理。
实验设计应包括工艺验证、热力学验证、制剂工艺的确定、包装工艺的确定等方面,具体分为以下几个环节:1.工艺验证:考量制剂工艺的可行性,验证所使用设备的关键参数,包括批次量、防刮器、可重复性等。
实验中应记录温度、压力和流量等信息。
2.热力学验证:根据不同的制剂工艺,设计冷冻干燥条件和试验方案。
一、冷冻干燥过程研究真空冷冻干燥是先将制品冻结到共晶点温度以下,使水分变成固态的冰,然后在适当的温度和真空度下,使冰升华为水蒸气。
再用真空系统的冷凝器(水汽凝结器)将水蒸气冷凝,从而获得干燥制品的技术。
该过程主要可分为:制品准备、预冻、一次干燥(升华干燥)、二次干燥(解吸干燥)和密封保存五个步骤。
1 产品预冻1.1 制品的玻璃化玻璃化的作用。
近年来,人们已经逐渐地认识到,凡是成功的低温保存,细胞内的水均以玻璃态的形式被固化,在胞内不出现晶态的冰。
玻璃化是指物质以非晶态形式存在的一种状态,其粘度极大,分子的能动性几乎为零,由于这种非晶体结构的扩散系数很低,故在这种结构中分子运动和分子变性反应非常微弱,不利的化学反应能够被抑制,从而提高被保存物质的稳定性。
玻璃化的获得。
在产品预冻时,只要降温速率足够快,且达到足够低的温度,大部分材料都能从液体过冷到玻璃态固体。
“足够快”的意思是在降温过程中迅速通过结晶区而不发生晶化,“足够低”指的是必须把温度降到玻璃化转变温度Tg以下。
对于具有一定初始浓度的细菌制品,其预冻过程一般通过“两步法”来完成。
第一步是以一般速率进行降温,让细胞外的溶液中产生冰,细胞内的水分通过细胞膜渗向胞外,胞内溶液的浓度逐渐提高;第二步是以较高速率进行降温,以实现胞内溶液的玻璃化。
此法又称“部分玻璃化法”。
当初始浓度为A的溶液(A点)从室温开始冷却时,随着温度的下降,溶液过冷到B点后将开始析出冰,结晶潜热的释放又使溶液局部温度升高。
溶液将沿着平衡的熔融线不断析出冰晶,冰晶周围剩余的未冻溶液随温度下降,浓度不断升高,一直下降到熔融线(Ta)与玻璃化转变曲线(Tg)的交点(D点)时,溶液中剩余的水分将不再结晶(称为不可冻水),此时的溶液达到最大冻结浓缩状,浓度较高,以非晶态的形式包围在冰晶周围,形成镶嵌着冰晶的玻璃体。
1.2降温速率与预冻温度预冻速度决定了制品体积大小、形状和成品最初晶格及其微孔的特性,其速度可控制在每分钟降温1℃左右。
冻干粉针剂设计说明书一、概述冻干粉针剂是一种常见的药物制剂,它通过冻干工艺将药物转化为粉末状,并将其装入注射器中,便于患者使用。
本说明书旨在详细介绍冻干粉针剂的设计原理、制备工艺以及质量控制等方面的内容。
二、设计原理冻干粉针剂的设计原理是将药物溶液冷冻成固体,在真空条件下进行冻干处理,以除去水分,从而得到稳定的粉末状药物。
冻干工艺能够保持药物的活性,并延长其保存期限,提高治疗效果。
三、制备工艺1. 药物溶液制备:首先,根据药物的性质选择合适的溶剂,将药物溶解于溶剂中,制备药物溶液。
要注意溶液的浓度和pH值的控制。
2. 冷冻:将药物溶液倒入冷冻容器中,使其迅速冷却成固体。
冷冻过程应该控制温度和速度,以确保药物冻结均匀,避免结晶。
3. 冻干处理:将冷冻的药物固体放入冻干器中,在真空条件下进行冻干处理。
冻干过程分为三个阶段:冷冻阶段、干燥阶段和干燥完成阶段。
4. 封装:冻干完成后,将冻干粉末装入注射器中,并密封好,以保护药物的稳定性和安全性。
四、质量控制1. 药物含量测定:通过药物含量测定方法,确定每支注射器中药物的含量是否符合要求。
常用的测定方法有高效液相色谱法、紫外分光光度法等。
2. 溶解度测定:检测药物冻干粉末在溶剂中的溶解度,以确定其溶解性能是否良好。
常用的测定方法有热重分析法、动态光散射法等。
3. 外观检查:对冻干粉针剂的外观进行检查,包括颜色、透明度、气泡等方面。
外观检查能够直观地判断药物的质量是否良好。
4. 稳定性研究:通过长期稳定性研究,确定冻干粉针剂的保存期限和储存条件。
稳定性研究包括药物含量变化、溶解度变化、外观变化等方面的评估。
五、注意事项1. 制备过程中要严格控制温度、湿度和真空度等参数,以确保药物质量的稳定性和一致性。
2. 制备过程中要注意避免药物的氧化、分解和聚集等不良反应的发生,以保持药物的活性。
3. 在储存和使用过程中,要避免暴露在光线、湿气和高温等不利环境下,以防止药物质量的降低。
冻干粉针剂的工艺流程图及环境要求生产工艺流程及环境区域划分示意图入 库冻干粉针剂认证检查的要点冻干粉针剂指在低温真空的环境下先将配制好的药液预冻然后升华干燥成粉末状态的一种制剂。
根据药品新版GMP原则要求,结合冻干粉针剂生产影响药品质量的关键环节,提出冻干粉针剂GMP检查要点,旨在督查药品生产企业质量保证体系和实际运行状态。
检查组须对如下内容按照药品GMP要求,逐一核实。
1、机构与人员a)主管生产和质量管理的企业负责人、生产管理和质量管理部门负责人均应具有医药或相关专业大专以上学历,并具有药品生产和质量管理经验,并履行其职责。
b)企业负责人和各级管理人员应定期接受药品管理法律法规培训。
c)质量检验、生产、维修保养、清洁人员应定期进行卫生和微生物学基础知识、洁净作业等方面的培训和考核,并具有实际操作技能。
2、厂房设施的管理要点及检查重点a)洁净区:我国《药品生产质量管理规范》对最终灭菌的无菌药品生产厂房洁净度级别的要求是:浓配或采用密闭系统的稀配应在100000级洁净区内进行;稀配、滤过、灌封、直接接触药品的包装材料的最终处理等操作应在10000级洁净区内进行;物料、中间品应经过物流缓冲间或传递柜进、出洁净区。
称量配料间如产尘应与洁净走廊呈相对负压,必要时设捕尘设施。
中国药典规定:微生物限度检查、无菌检查应在100级或10000级背景下的局部100级区内进行,并与生产区分开。
微生物限度检查与无菌检查用的实验室和空气净化系统最好彼此分开,以尽可能减少对无菌检查的干扰。
b)空气净化系统应能确保洁净区的洁净度级别、温湿度、压差等符合生产工艺要求并经过验证。
初效、中效过滤器应明确清洗/更换周期,高效过滤器应定期检测其完整性,如有泄漏或阻塞应及时更换。
空气净化系统应每天24小时运行,停用后再次运行应进行清洁、消毒并经过再验证,符合要求的方可开始生产。
c)与产品直接接触的压缩空气、氮气、二氧化碳等辅助设施这些气体因与产品直接接触,不得对产品带来污染。
应对系统进行验证。
PQ测试项目包括洁净度级别、含水量、含油量等。
d)注射用水(WFI)系统以纯化水为原水,经多效蒸馏制得。
制水系统应能提供足够量的符合质量要求的注射用水。
注射用水制备和分配系统材质应无毒、耐腐蚀,设计和安装应避免死角和盲管;储罐应密闭,并安装有经完整性检查合格的无菌级别的疏水性过滤器,设有保温装置;管道通过卡箍连接,有一定的倾斜度,最低位出水,阀门用隔膜阀而不是球阀;注射用水的储存应采用80o C以上保温或65o C以上保温循环,应有温度控制和指示装置;设有必要的取样口包括注射用水储罐的入水口、总出水口和总回水口,并便于取样;储罐、增压泵、管道可以排空便于清洁、消毒或灭菌。
e)纯蒸汽系统纯蒸汽通常用于与产品接触物品的灭菌及产品的最终灭菌,质量应符合要求,纯蒸汽系统应经过验证并定期监测。
3、设备a)配制系统称量用秤的精度应符合物料的称量要求,并定期校验,贴有合格标志。
配制罐体积应与生产批量相匹配,材质应符合要求,禁用铁类容器或露铁搪瓷桶,配有搅拌、降温等装置,可定容。
配制罐宜自带称量系统或液位电极、或液位标尺,如采用玻璃液位管应能拆卸,以便清洗。
因配制需要在低于30℃、 CO2饱和的注射用水溶解原辅料,应有有效的控温装置,CO2输送管道终端应连有0.45 μm或精度更高的疏水性过滤器。
配有粗滤所需要的0.45μm 过滤器。
配有终端过滤所需要的2个0.22μm过滤器及过滤器完整性测试设备。
过滤器的安装应符合工艺要求,一个0.22μm的过滤器应可能接近灌封机(欧盟无菌药品附录要求),与产品直接接触的过滤器材质应不得吸附药液组分和释放异物(应有验证数据支持)。
反复使用的过滤芯应灭菌干燥后保存,并规定有灭菌有效期和使用次数。
配制系统宜采用密闭系统,并配有自动清洗、消毒或灭菌装置。
管道通过卡箍连接,使用结束应能排空。
b)洗瓶与灭菌洗瓶机:应安装在100000级洁净区。
洗安瓿能力应能满足工艺需要,洗净后安瓿的清洁度符合要求。
隧道烘箱:应安装在100000级洁净区,分为预热、高温、冷却三个部分,应有温度记录装置,应有传送速度显示仪,温度控制仪与温度记录装置应分开。
传送带不得穿越十万级与万级区域,应分段。
定期检测隧道烘箱内洁净度和风速,如:尘埃粒子、沉降菌等数据。
高效过滤器应定期或根据两端压差的变化情况进行更换,更换后应进行检漏试验。
c)液体半加塞灌装机组联动线灌封系统:应安装在10000级洁净区,能有效控制装量,灌装过程中药液无溅壁现象,安瓿封口完好。
灌装头、灌装管道等灌装器具应按规定的程序清洁、消毒或灭菌。
灌封速度,达到产量要求。
灌装量准确性,应符合中国药典规定,以容积法检查,装量误差≤2.5%半加胶塞正确成功率≥99%(对半加塞正确性检查用目测法,认可标准是无压斜和涤槽胶塞槽外露);胶塞定向失误率≤3‰,缺瓶不灌装动作完成率100%,工作时的挤瓶破损率<1‰。
灌封后的澄明度测试,灌封后的澄明度≥96%;灌封时的不溶性粒子的测试,应符合中国药典中。
控制与显示关键参数的仪表如:温度、传送速度应经过定期校验,并贴有合格校验标签。
d)文件每台主要设备都应建立设备使用标准操作程序(SOP)、清洁、消毒或灭菌标准操作程序、维护保养标准操作程序、使用记录和设备档案。
4、物料的管理要点及检查重点a)物料的采购应由具有产品专业知识的人员负责,从企业质量审计合格的供应商处采购。
内包装材料为低硼硅玻璃安瓿,应符合国家药用包装容器(材料)标准,标准号:国家药用包装容器(材料)标准(试行)YBB00332002。
b)原辅料的检验、贮存及分发物料应保存于企业规定的适宜的环境中并有明确的标识,质控部门应按批取样、检验、签发。
取样应具有代表性,样品应按企业内控标准进行检验。
原辅料应经质量部门签发后在有效期内使用。
不合格的原辅料不得用于生产。
c)包装材料低硼硅玻璃安瓿经检验合格后方可投入使用。
药品标签和说明书应与药品监督管理部门批准的内容、式样、文字相一致,经企业质量管理部门校对无误后印制、发放、使用。
标签发放、使用、销毁应有记录。
d)不合格品不合格物料、产品应专区存放,应有易于识别的明显标志,并按有关规定及时处理,并有记录。
对不合格产品应查明原因并采取必要的纠正措施。
e)退回产品、收回产品应按相关程序处理并作适当记录。
5、验证的管理要点及检查重点a)HVAC系统及洁净室的验证新车间应进行前验证,包括IQ、OQ和PQ,车间运行一定时间或停止运行后再次使用前应进行再验证。
验证测试项目应重点检查洁净度级别(尘埃粒子、微生物)、高效过滤器完整性(DOP)、压差、温、湿度(应能满足生产工艺要求)、局部100级区的气流流型、洁净室的清洁及消毒措施等。
b)压缩空气、氮气、二氧化碳等辅助系统的验证新系统应进行前验证,包括IQ、OQ和PQ,系统运行一定时间后应进行再验证。
气体储存及分配系统材质应无毒,并设终端过滤器。
验证测试项目应包括洁净度级别(尘埃粒子、微生物)、含水量、含油量等。
c)注射用水系统的验证新的注射用水系统应进行前验证,包括IQ、OQ和PQ。
注射用水储罐和分配系统选用材质应无毒、耐腐蚀,管道的设计和安装应避免了死角、盲管,储罐应安装有疏水性过滤器,储罐和管道应能排空,注射用水及分配系统的保温效果应能达到设计要求(80o C以上保温或65o C以上保温循环)。
注射用水质量应不低于中国药典(2005年版)注射用水质量标准,验证测试时间应不少于3周,并应监测季节变化对水质的影响。
企业应根据验证结果规定注射用水系统的清洗、灭菌周期。
企业应制定注射用水系统监测计划,包括对微生物、内毒素、理化指标,并按计划对水质进行监测并有完整记录,鼓励企业做趋势分析、年度回顾。
d)液体半加塞灌装机组的验证新系统应进行前验证,包括IQ、OQ和PQ,系统运行一定时间后应进行再验证。
洗瓶机清洗安瓿后的洁净程度(清洗后往安瓿中加入注射用水,振摇后检查澄明度和不溶性颗粒)。
隧道烘箱高效过滤器的完整性和隧道烘箱内洁净度级别、隧道烘箱空载热分布、负载热穿透试验以及微生物挑战试验。
灌装机装量准确性、灌装后安瓿熔封效果等。
e)湿热灭菌工艺的验证中国药典(2005年版)要求,对热稳定的产品,可采用过度杀灭法,其无菌保证水平(SAL)应≤10-12,热稳定较差产品的标准灭菌时间F0一般不低于8分钟,此情况下,应在生产全过程中,对产品中污染的微生物严加监控,并采取各种措施降低微生物污染水平,确保被灭菌产品达到无菌保证要求。
在注射液生产中,产品的最终灭菌和与产品直接接触的器具如过滤器的灭菌等采用湿热灭菌。
该产品最终灭菌的工艺参数为121°C 20分钟。
进行灭菌验证时,被灭菌物品的数量和装载方式、灭菌温度和时间或F0必须在验证方案中明确规定,至少进行3次空载热分布试验,每种装载方式都至少进行3次负载热穿透试验,并选用合适的生物指示剂。
空载热分布试验和负载热穿透试验至少用12个热电偶,应有热电偶分布示意图,热电偶在每次使用前后均应进行校正。
121°C蒸汽灭菌通常选用中嗜热脂肪芽孢杆菌作为生物指示剂。
验证过程中应考虑灭菌参数的上限和下限,保证灭菌效果和产品质量。
f)工艺验证工艺验证应在洁净室系统、压缩空气及氮气系统、工艺用水系统、热力灭菌系统、检测方法等验证合格且人员经相关培训考核合格后进行。
应连续3批产品验证合格。
验证的批量应与工艺规程规定的相同,关键工艺控制参数如配制工序中的pH值、温度、搅拌时间、隧道烘箱灭菌温度及安瓿输送带速度、灌装工序中的装量、最终灭菌的温度及时间等是应在工艺规程规定的范围之内。
采集的用于检验的样品应足够,能反映整批产品的质量。
如检查安瓿是否清洗干净时,应在清洗安瓿的前、中、后期取样对安瓿的洁净度进行检查;检查装量时,应在灌装的前、中、后期采集了足够的样品并分别进行检验;成品应按灭菌柜次取样并分别作无菌试验等。
工艺验证应包括最差条件,如从配制到灌装或灭菌的时间间隔要求、灌装期间进入灌装间人员的数量、设备出现故障时进行必要的检修等。
灌装期间应对灌装间进行微生物监测,监测结果应作为产品放行审核的一部分。
变更控制和偏差管理应符合规定,对出现的偏差应进行了调查和记录等。
g)清洁验证不同产品共用生产设备,或产品不稳定生成了降解产物,或清洁过程引入了外来物质如清洁剂等,应进行清洁验证。
清洁验证是对某一具体的清洁程序进行验证。
重点检查清洁程序的可操作性、清洁过程中是否引入了外来物质、允许残留量计算的依据、化学残留量检测方法的灵敏度、是否考虑了所有共用设备、管道中的残留等。
清洁验证判断标准应包括目测、漂洗液取样检验和棉签擦拭取样检验。
棉签擦拭取样检验应做回收率试验作为取样、检测方法的依据。