数据分析文献阅读_测序深度和覆盖度
- 格式:ppt
- 大小:39.00 KB
- 文档页数:24

基因组测序实验报告一、实验背景随着生命科学的快速发展,基因组测序技术已经成为研究生物遗传信息的重要手段。
通过对基因组的测序,可以深入了解生物的基因组成、遗传变异、基因功能以及与疾病的关系等。
本次实验旨在对_____样本进行基因组测序,以获取其详细的遗传信息。
二、实验目的1、掌握基因组测序的基本原理和实验流程。
2、对_____样本进行全基因组测序,获得高质量的测序数据。
3、分析测序数据,查找可能存在的基因突变和遗传变异。
三、实验材料与方法(一)实验材料1、样本来源:_____2、试剂与仪器:DNA 提取试剂盒测序试剂盒测序仪离心机移液器等(二)实验方法1、 DNA 提取按照 DNA 提取试剂盒的说明书,从_____样本中提取高质量的基因组 DNA。
对提取的 DNA 进行浓度和纯度检测,确保其质量符合测序要求。
2、文库构建将提取的 DNA 进行片段化处理,使其大小适合测序。
对片段化的 DNA 进行末端修复和加接头等操作,构建测序文库。
3、测序将构建好的测序文库加载到测序仪上,进行测序反应。
选择合适的测序模式和参数,以获得高质量的测序数据。
4、数据处理与分析对测序得到的原始数据进行质量评估和过滤,去除低质量的数据。
使用专业的生物信息学软件对处理后的数据进行比对、组装和变异检测等分析。
四、实验结果(一)测序数据质量评估1、测序深度:平均测序深度达到_____X,覆盖度良好。
2、碱基质量:碱基质量值的分布符合预期,大部分碱基的质量值在 Q30 以上。
(二)基因组装结果成功组装出_____样本的基因组序列,与已知的参考基因组相比,具有较高的一致性。
(三)变异检测结果1、单核苷酸多态性(SNP):共检测到_____个 SNP 位点,分布在不同的染色体上。
2、插入缺失(InDel):检测到_____个 InDel 变异,其长度和位置分布具有一定的特征。
(四)功能注释与分析对检测到的变异进行功能注释,发现其中一些变异可能与_____疾病的发生发展相关。

1G=1024M测序深度是指测序得到的总碱基数与待测基因组大小的比值.假设一个基因大小为2M,测序深度为10X,那么获得的总数据量为20M.测序深度=总数据量20M/基因组大小2M=10X覆盖度是指测序获得的序列占整个基因组的比例.由于基因组中的高GC、重复序列等复杂结构的存在,测序最终拼接组装获得的序列往往无法覆盖有所的区域,这部分没有获得的区域就称为Gap.例如一个细菌基因组测序,覆盖度是98%,那么还有2%的序列区域是没有通过测序获得的.核苷酸多态性位点SNP,插入缺失位点InDel,Insertion/Deletion、结构变异位点SV,StructureVariation位点.SBC可以协助客户,通过手段,分析不同间的结构差异,同时完成注释.技术路线提取基因组DNA,利用Covaris进行随机打断,电泳回收所需长度的DNA片段0.2~5Kb,加上接头,进行cluster制备Solexa或E-PCRSOLiD,最后利用Paired-EndSolexa或者Mate-PairSOLiD的方法对插入片段进行重测序.图1-1,以SOLiD为例,说明整个实验方案.2、外显子测序也称目标组捕获,是指利用序列捕获技术将全基因组外显子区域DNA捕捉并富集后进行高通量测序的基因组分析方法.是一种选择基因组的的高效策略,外显子测序相对于基因组重测序成本较低,对研究已知基因的SNP、Indel等具有较大的优势.外显子expressedregion是真核生物基因的一部分,它在剪接Splicing后仍会被保存下来,并可在蛋白质生物合成过程中被表达为蛋白质.外显子是最后出现在成熟RNA中的基因序列,又称表达序列.既存在于最初的产物中,也存在于成熟的RNA分子中的核苷酸序列.在人类基因中大约有180,000,占人类基因组的1%,约30MB.。
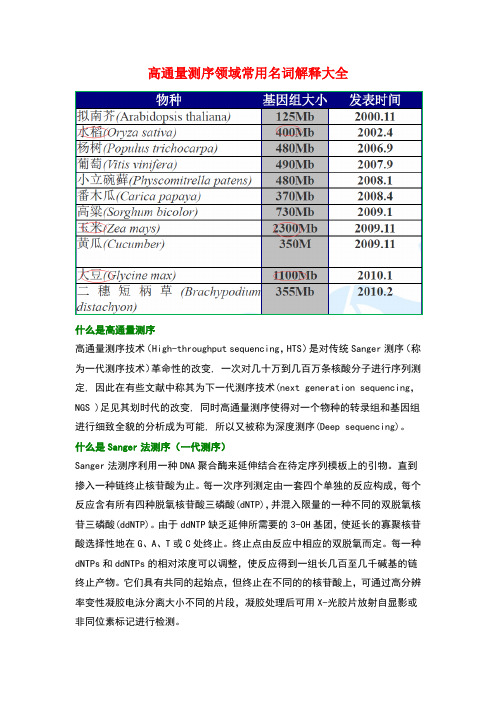
高通量测序领域常用名词解释大全什么是高通量测序高通量测序技术(High-throughput sequencing,HTS)是对传统Sanger测序(称为一代测序技术)革命性的改变, 一次对几十万到几百万条核酸分子进行序列测定, 因此在有些文献中称其为下一代测序技术(next generation sequencing,NGS )足见其划时代的改变, 同时高通量测序使得对一个物种的转录组和基因组进行细致全貌的分析成为可能, 所以又被称为深度测序(Deep sequencing)。
什么是Sanger法测序(一代测序)Sanger法测序利用一种DNA聚合酶来延伸结合在待定序列模板上的引物。
直到掺入一种链终止核苷酸为止。
每一次序列测定由一套四个单独的反应构成,每个反应含有所有四种脱氧核苷酸三磷酸(dNTP),并混入限量的一种不同的双脱氧核苷三磷酸(ddNTP)。
由于ddNTP缺乏延伸所需要的3-OH基团,使延长的寡聚核苷酸选择性地在G、A、T或C处终止。
终止点由反应中相应的双脱氧而定。
每一种dNTPs和ddNTPs的相对浓度可以调整,使反应得到一组长几百至几千碱基的链终止产物。
它们具有共同的起始点,但终止在不同的的核苷酸上,可通过高分辨率变性凝胶电泳分离大小不同的片段,凝胶处理后可用X-光胶片放射自显影或非同位素标记进行检测。
什么是基因组重测序(Genome Re-sequencing)全基因组重测序是对基因组序列已知的个体进行基因组测序,并在个体或群体水平上进行差异性分析的方法。
随着基因组测序成本的不断降低,人类疾病的致病突变研究由外显子区域扩大到全基因组范围。
通过构建不同长度的插入片段文库和短序列、双末端测序相结合的策略进行高通量测序,实现在全基因组水平上检测疾病关联的常见、低频、甚至是罕见的突变位点,以及结构变异等,具有重大的科研和产业价值。
什么是de novo测序de novo测序也称为从头测序:其不需要任何现有的序列资料就可以对某个物种进行测序,利用生物信息学分析手段对序列进行拼接,组装,从而获得该物种的基因组图谱。

单细胞测序基因数和数据量关系
单细胞测序(Single-Cell Sequencing)是一种用于研究单个细胞基因表达的高通量技术。
基因数和数据量之间的关系在单细胞测序中是一个重要的考虑因素。
以下是一些相关的考虑和观点:
1.基因数与数据量:
o单细胞测序的数据量通常与测序深度(每个细胞的平均读数)和测序覆盖度(涵盖基因组中的多少
部分)有关。
o更高的测序深度和覆盖度通常可以检测到更多的基因,尤其是在低表达水平的基因中。
o由于单细胞测序的数据量较大,通常需要处理大量的基因表达矩阵,这可能对数据存储、处理和分
析带来挑战。
2.细胞数和数据量:
o单细胞测序通常包括对多个细胞的测序,细胞数目的增加将显著增加整体的数据量。
o更多的细胞数可能提供更全面的单细胞分析,但也增加了数据处理和存储的复杂性。
3.样本类型和实验设计:
o样本类型和实验设计的选择也会影响单细胞测序的数据量需求。
例如,研究者可能在相同数量的细
胞中选择深度测序较高的样本,以便更深入地探
索细胞的基因表达。
4.技术平台:
o使用不同的单细胞测序技术平台(如10x Genomics、Drop-seq等)可能会对数据量产生影
响。
一些平台可能更适合高通量测序,而另一些则
更适合特定实验设计或预算。
总体而言,单细胞测序的数据量需求是一个平衡问题,需要根据具体研究目的、预算和实验设计来确定。
在进行单细胞测序实验时,研究者应该仔细考虑数据量的要求,以确保能够满足科学问题的需要。

全基因组测序指南(一):名词解释Barcode条形码,因为在测序过程中往往是多个样本一起测序,所以Barcode的作用就是区分测序产生序列到底是来源于哪一个个体;另外,10X Genomics在测序过程中也用到了Barcode,其作用是区分测序产生的序列是否来源于同一个大片段,更加有利于组装成更高质量的基因组。
BAC细菌人工染色体(Bacterial artificial chromosome),可以构建超长片段的DNA序列,在基因组组装过程中有助于组装更高质量的基因组,也可用于基因组的组装质量评估。
Contig拼接软件基于reads之间的重叠(overlap)区,拼接获得的没有gap的序列称为Contig(重叠群)。
与之对应的一个概念是Scaffold:是指将获得的Contig根据大片段文库的Pair-end关系,将Contig进一步组装成更长的序列。
ContigN50衡量Contig的一个指标。
即将所有的Contig长度相加,获得一个Contig总长度,然后将所有的Contigs按照从长到短进行排序,将Contig按照这个顺序依次相加,当相加的长度达到Contig为总长度的一半时,最后一个加上的Contig长度即为ContigN50。
ContigN90概念与N50的类似,即当相加的长度达到Contig总长度的90%时,最后一个加上的Contig长度即为Contig N90。
N50或者N90是衡量基因组质量的一个重要指标。
ScaffoldN50衡量Scaffold的一个指标。
即将所有的Scaffold长度相加,获得一个Scaffold总长度,然后将所有的Scaffold按照从长到短进行排序,将Scaffold按照这个顺序依次相加,当相加的长度达到Scaffold总长度的一半时,最后一个加上的Scaffold长度即为Scaffold N50。
ScaffoldN90的概念与N50的类似,即当相加的长度达到Scaffold总长度的90%时,最后一个加上的Scaffold长度即为Scaffold N90。

全基因组重测序数据分析1. 简介(Introduction)通过高通量测序识别发现de novo的somatic和germ line 突变,结构变异-SNV,包括重排突变(deletioin, duplication 以及copy number variation)以及SNP的座位;针对重排突变和SNP的功能性进行综合分析;我们将分析基因功能(包括miRNA),重组率(Recombination)情况,杂合性缺失(LOH)以及进化选择与mutation之间的关系;以及这些关系将怎样使得在disease(cancer)genome中的mutation产生对应的易感机制和功能。
我们将在基因组学以及比较基因组学,群体遗传学综合层面上深入探索疾病基因组和癌症基因组。
实验设计与样本(1)Case-Control 对照组设计;(2)家庭成员组设计:父母-子女组(4人、3人组或多人);初级数据分析1.数据量产出:总碱基数量、Total Mapping Reads、Uniquely Mapping Reads统计,测序深度分析。
2.一致性序列组装:与参考基因组序列(Reference genome sequence)的比对分析,利用贝叶斯统计模型检测出每个碱基位点的最大可能性基因型,并组装出该个体基因组的一致序列。
3.SNP检测及在基因组中的分布:提取全基因组中所有多态性位点,结合质量值、测序深度、重复性等因素作进一步的过滤筛选,最终得到可信度高的SNP数据集。
并根据参考基因组信息对检测到的变异进行注释。
4.InDel检测及在基因组的分布: 在进行mapping的过程中,进行容gap的比对并检测可信的short InDel。
在检测过程中,gap的长度为1~5个碱基。
对于每个InDel的检测,至少需要3个Paired-End序列的支持。
5.Structure Variation检测及在基因组中的分布: 能够检测到的结构变异类型主要有:插入、缺失、复制、倒位、易位等。
什么是高通量测序?高通量测序技术(High-throughput sequencing,HTS)是对传统Sanger测序(称为一代测序技术)革命性的改变, 一次对几十万到几百万条核酸分子进行序列测定, 因此在有些文献中称其为下一代测序技术(next generation sequencing,NGS )足见其划时代的改变, 同时高通量测序使得对一个物种的转录组和基因组进行细致全貌的分析成为可能, 所以又被称为深度测序(Deep sequencing)。
什么是Sanger法测序(一代测序)Sanger法测序利用一种DNA聚合酶来延伸结合在待定序列模板上的引物。
直到掺入一种链终止核苷酸为止。
每一次序列测定由一套四个单独的反应构成,每个反应含有所有四种脱氧核苷酸三磷酸(dNTP),并混入限量的一种不同的双脱氧核苷三磷酸(ddNTP)。
由于ddNTP缺乏延伸所需要的3-OH基团,使延长的寡聚核苷酸选择性地在G、A、T或C处终止。
终止点由反应中相应的双脱氧而定。
每一种dNTPs和ddNTPs的相对浓度可以调整,使反应得到一组长几百至几千碱基的链终止产物。
它们具有共同的起始点,但终止在不同的的核苷酸上,可通过高分辨率变性凝胶电泳分离大小不同的片段,凝胶处理后可用X-光胶片放射自显影或非同位素标记进行检测。
什么是基因组重测序(Genome Re-sequencing)全基因组重测序是对基因组序列已知的个体进行基因组测序,并在个体或群体水平上进行差异性分析的方法。
随着基因组测序成本的不断降低,人类疾病的致病突变研究由外显子区域扩大到全基因组范围。
通过构建不同长度的插入片段文库和短序列、双末端测序相结合的策略进行高通量测序,实现在全基因组水平上检测疾病关联的常见、低频、甚至是罕见的突变位点,以及结构变异等,具有重大的科研和产业价值。
什么是de novo测序de novo测序也称为从头测序:其不需要任何现有的序列资料就可以对某个物种进行测序,利用生物信息学分析手段对序列进行拼接,组装,从而获得该物种的基因组图谱。
高通量测序领域常用名词解释大全什么是高通量测序?高通量测序技术(High-throughput sequencin,g HTS)是对传统Sanger测序(称为一代测序技术)革命性的改变, 一次对几十万到几百万条核酸分子进行序列测定, 因此在有些文献中称其为下一代测序技术(next generation sequencin,g NGS )足见其划时代的改变, 同时高通量测序使得对一个物种的转录组和基因组进行细致全貌的分析成为可能, 所以又被称为深度测序(Deep sequencing。
)什么是Sanger法测序(一代测序)Sanger法测序利用一种DNA 聚合酶来延伸结合在待定序列模板上的引物。
直到掺入一种链终止核苷酸为止。
每一次序列测定由一套四个单独的反应构成,每个反应含有所有四种脱氧核苷酸三磷酸(dNTP),并混入限量的一种不同的双脱氧核苷三磷酸(ddNTP)。
由于ddNTP 缺乏延伸所需要的3-OH 基团,使延长的寡聚核苷酸选择性地在G、A、T 或C 处终止。
终止点由反应中相应的双脱氧而定。
每一种dNTPs 和ddNTPs 的相对浓度可以调整,使反应得到一组长几百至几千碱基的链终止产物。
它们具有共同的起始点,但终止在不同的的核苷酸上,可通过高分辨率变性凝胶电泳分离大小不同的片段,凝胶处理后可用X-光胶片放射自显影或非同位素标记进行检测。
什么是基因组重测序(Genome Re-sequencin)g全基因组重测序是对基因组序列已知的个体进行基因组测序,并在个体或群体水平上进行差异性分析的方法。
随着基因组测序成本的不断降低,人类疾病的致病突变研究由外显子区域扩大到全基因组范围。
通过构建不同长度的插入片段文库和短序列、双末端测序相结合的策略进行高通量测序,实现在全基因组水平上检测疾病关联的常见、低频、甚至是罕见的突变位点,以及结构变异等,具有重大的科研和产业价值。
什么是de novo 测序de novo 测序也称为从头测序:其不需要任何现有的序列资料就可以对某个物种进行测序,利用生物信息学分析手段对序列进行拼接,组装,从而获得该物种的基因组图谱。
测序原始数据参数测序原始数据(Raw Data)通常指的是测序仪器直接产生的未经处理的数据。
这些数据通常以fastq文件格式存储,记录了测序样本的碱基序列信息以及与之相关的质量评分。
在处理这些原始数据时,通常会涉及一些参数设置,以下是一些常见的参数及其说明:1. 测序平台与试剂:不同的测序平台(如Illumina、PacBio、Oxford Nanopore等)和试剂会影响数据的质量和产出。
因此,了解所使用的测序平台和试剂对于数据解读至关重要。
2. 测序深度:测序深度指的是测序覆盖的基因组区域的倍数。
较高的测序深度可以提高数据的准确性和可靠性,但也会增加成本和计算负担。
3. 质量评分系统:测序数据中的每个碱基通常都会有一个与之相关的质量评分,用于表示该碱基的测序可靠性。
不同的测序平台和软件可能使用不同的质量评分系统,如Illumina的ASCII编码或Sanger 的Phred编码。
4. 数据过滤:在原始数据中,可能存在一些低质量的序列或碱基,这些数据在后续分析中可能会产生干扰。
因此,通常需要对原始数据进行过滤,去除低质量的序列或碱基。
过滤的标准可能包括序列长度、平均质量评分等。
5. 数据拆分与合并:对于双端测序(Paired-End Sequencing)产生的数据,需要将两个端的数据进行拆分和合并。
拆分时需要根据测序引物的序列和位置信息来确定每个端的数据范围;合并时则需要将两个端的数据按照基因组的位置信息进行拼接。
6. 数据压缩与存储:由于原始数据通常较大,为了方便存储和传输,可能需要对数据进行压缩。
常用的压缩格式包括gzip(.gz)和bzip2(.bz2)等。
了解这些参数对于正确处理和解读测序原始数据至关重要。
在实际操作中,建议根据具体的测序平台、试剂和实验需求来选择合适的参数设置。
高通量测序基础知识简介陆桂什么是高通量测序?高通量测序技术(High-throughput sequencing,HTS)是对传统Sanger测序(称为一代测序技术)革命性的改变,一次对几十万到几百万条核酸分子进行序列测定, 因此在有些文献中称其为下一代测序技术(next generation sequencing,NGS )足见其划时代的改变, 同时高通量测序使得对一个物种的转录组和基因组进行细致全貌的分析成为可能, 所以又被称为深度测序(Deep sequencing)。
什么是Sanger法测序(一代测序)Sanger法测序利用一种DNA聚合酶来延伸结合在待定序列模板上的引物。
直到掺入一种链终止核苷酸为止。
每一次序列测定由一套四个单独的反应构成,每个反应含有所有四种脱氧核苷酸三磷酸(dNTP),并混入限量的一种不同的双脱氧核苷三磷酸(ddNTP)。
由于ddNTP缺乏延伸所需要的3-OH基团,使延长的寡聚核苷酸选择性地在G、A、T或C处终止。
终止点由反应中相应的双脱氧而定。
每一种dNTPs和ddNTPs的相对浓度可以调整,使反应得到一组长几百至几千碱基的链终止产物。
它们具有共同的起始点,但终止在不同的的核苷酸上,可通过高分辨率变性凝胶电泳分离大小不同的片段,凝胶处理后可用X-光胶片放射自显影或非同位素标记进行检测。
什么是基因组重测序(Genome Re-sequencing)全基因组重测序是对基因组序列已知的个体进行基因组测序,并在个体或群体水平上进行差异性分析的方法。
随着基因组测序成本的不断降低,人类疾病的致病突变研究由外显子区域扩大到全基因组范围。
通过构建不同长度的插入片段文库和短序列、双末端测序相结合的策略进行高通量测序,实现在全基因组水平上检测疾病关联的常见、低频、甚至是罕见的突变位点,以及结构变异等,具有重大的科研和产业价值。
什么是de novo测序de novo测序也称为从头测序:其不需要任何现有的序列资料就可以对某个物种进行测序,利用生物信息学分析手段对序列进行拼接,组装,从而获得该物种的基因组图谱。