食品理化检验方法标准.
- 格式:ppt
- 大小:1.30 MB
- 文档页数:58

蛋白质检测1、试样处理称取试样0.5g(精确至0.001g),用称量纸包住,连同称量纸移入消化管内,加入0.5g硫酸铜、4.5g硫酸钾和10ml硫酸,放到红外智能消化炉上消化。
消化完取出冷却,加入40ml水,于凯氏定氮仪(使用前加入40%氢氧化钠溶液,2%硼酸溶液,水)上实现自动加液、蒸馏。
试样取量只针对本司生产的奶片糖。
红外智能消化炉使用步骤:消化管放到消化炉上,盖上消化盖,打开水龙头,插上插头,打开电源跟启动器,双手同时按住“SET”跟“A/M”,跳到程序10之后按“SET”,再按“△”调到程序3,最后红外智能消化炉自行消化。
使用结束后关闭启动器、电源,拔掉插头。
凯氏定氮仪使用步骤:先打开冷却水,插上插头,打开电源,按“+”键调至程序1,按“启动”键开始进行测试。
仪器使用结束后,并把碱管放置在清水桶中,按“-”调至程序0,按“启动”键清洗3-4次,清洗完碱管放回碱桶内,关闭电源,拔掉插头,关闭水龙头,擦拭凯氏定氮仪。
2、滴定向接受瓶内加入10滴溴甲酚绿一甲基红指示剂,用0.1mol/L盐酸标准溶液滴定至终点并记录滴定数据,终点颜色为灰红色。
同时做试剂空白。
3、结果计算试样中蛋白质含量按下式计算:10025.6)(0140.01⨯⨯-⨯⨯=mVVcX式中:X ——试样中蛋白质含量,单位为克每百克(g/100g);0.0140 ——1.0ml盐酸标准滴定溶液相当的氮质量,单位为克(g);c ——盐酸标准滴定溶液浓度,单位为摩尔每升(mol/L);V1——试液消耗盐酸标准滴定液的体积,单位为毫升(ml);V——试剂空白消耗盐酸标准滴定液的体积,单位为毫升(ml);m ——试样的质量,单位为克(g);6.25 ——蛋白质折算系数;100 ——换算系数。
蛋白质含量≥1g/100g是,结果保留三位有效数字;蛋白质含量<1g/100g 时,结果保留两位有效数字。
在重复条件下获得的两次独立测定结果的绝对差值不得超过算术平均值的10%。



密度瓶法:测定时用同一已知质量的密度瓶,先后盛满待测溶液和纯水,在同一温度下称重,根据空瓶的质量、纯水和待测溶液的质量计算待测溶液的密度。
方法:先将密度瓶洗净→干燥后准确称空瓶质量→装满样液加瓶塞→浸入20℃恒温水浴半小时→用滤纸条擦干外壁→加盖后称重→试样倾出→洗净加满纯水加瓶塞→重复上法。
适用于测定样品较少的液态食品相对密度计法:将相对密度计洗净擦干→缓缓放入盛有待测试液的量筒→静止后轻轻下按→待其自然上升,静止后读数。
食品样品的采集方法:随机采样(最常用)和代表性取样。
固态食品:采样件数=根号下总件数/2,四分法对角取样。
液态半固态:混合后虹吸法分上中下三层采样。
组成不均:采取不同部位。
含毒掺伪:采取含毒物或掺伪最多的部位无机化处理:湿消化法,干灰化法。
敞口瓶消化法(最常用):凯氏烧瓶或硬质锥形瓶。
凯氏烧瓶长颈可以起到回流作用,减少酸的挥发。
消化前加入样品和消化试剂,倾斜45℃,用电炉或电热板加热,直至消化完全。
应在通风橱内进行。
回流消化法:测定含有挥发性成分的食品样品。
装置上端连接冷凝器,可使挥发性成分随同酸雾冷凝流回反应瓶内。
冷消化法:低温消化法,置于室温或37~40℃烘箱内,放置过夜。
可避免易挥发的元素的挥发损失,仅适用于有机物较少的样品。
固相萃取SPE:基于液相色谱分离原理的样品制备技术,就是柱色谱分离方法。
在小柱中填充适当的固定相制成固相萃取柱,当样品液通过SPE小柱时,待测成分被吸留,用适当的溶液洗涤出去样品基体或杂质,然后用一种选择性的溶液将待测组分洗脱,达到分离、净化和浓缩的目的。
分为吸附、分配、离子交换、凝胶过滤、螯合亲和固相萃取。
超临界流体的萃取SFE:萃取剂为超临界流体,超临界流体是一类只能在温度和压力超过临界点才能存在的物态。
密度较大,与液体相近,可用作溶剂溶解其他物质;粘度较小,与气态相近,传质速度很快,而且表面张力小,很容易渗透进入固体样品内。
高效、快速。
常用的超临界溶剂为二氧化碳。
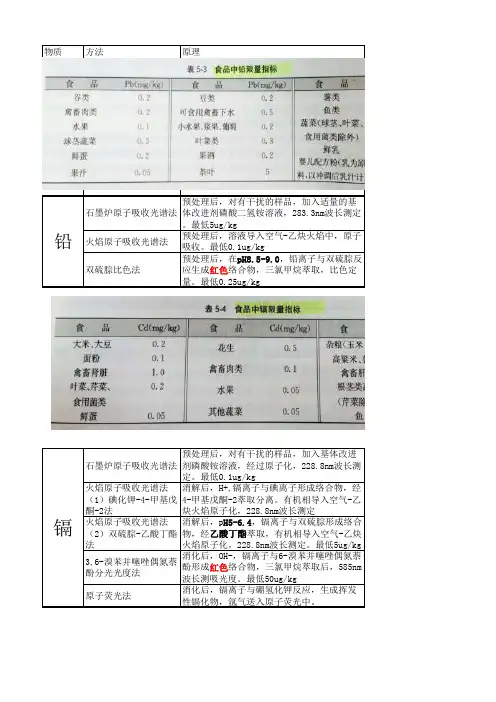
物质方法原理石墨炉原子吸收光谱法预处理后,对有干扰的样品,加入适量的基体改进剂磷酸二氢铵溶液,283.3nm波长测定。
最低5ug/kg火焰原子吸收光谱法预处理后,溶液导入空气-乙炔火焰中,原子吸收。
最低0.1ug/kg双硫腙比色法预处理后,在pH8.5-9.0,铅离子与双硫腙反应生成红色络合物,三氯甲烷萃取,比色定量。
最低0.25ug/kg石墨炉原子吸收光谱法预处理后,对有干扰的样品,加入基体改进剂磷酸铵溶液,经过原子化,228.8nm波长测定。
最低0.1ug/kg 火焰原子吸收光谱法(1)碘化钾-4-甲基戊酮-2法消解后,H+,镉离子与碘离子形成络合物,经4-甲基戊酮-2萃取分离。
有机相导入空气-乙炔火焰原子化,228.8nm波长测定火焰原子吸收光谱法(2)双硫腙-乙酸丁酯法消解后,p H5-6.4,镉离子与双硫腙形成络合物,经乙酸丁酯萃取,有机相导入空气-乙炔火焰原子化,228.8nm波长测定。
最低5ug/kg 3,6-溴苯并噻唑偶氮萘酚分光光度法消化后,OH-,镉离子与6-溴苯并噻唑偶氮萘酚形成红色络合物,三氯甲烷萃取后,585nm 波长测吸光度。
最低50ug/kg 原子荧光法消化后,镉离子与硼氢化钾反应,生成挥发性镉化物,氩气送入原子荧光中。
铅镉原子荧光法经高压消解或微波消解后,汞离子被还原成汞原子。
最低0.03ug/L,标曲最佳线性范围0~60ug/L 冷原子吸收光谱法经高压消解或微波消解后,汞离子被还原成汞原子,测汞仪测定原子吸收强度。
最低:压力消解法:0.4ug/kg,其他10ug/kg 双硫腙分光光度法消化后,汞离子在酸性溶液(pH1~2)与双硫腙生成橙红络合物,三氯甲烷萃取,测定波长510nm 。
最低25ug/kg GC(酸提取疏基棉法)试样中的甲基汞,用氯化钠研磨后加入含Cu 2+的盐酸(1:11),Cu 2+与组织中结合的CH 3Hg交换,萃取后,经离心过滤,将上清液调至一定酸度,用疏基棉吸附,再用盐酸(1:5)洗脱,最后以苯萃取甲基汞,ECD-GC测定冷原子吸收法(酸提取疏基棉法)同上,用测汞仪测定铝-铬天青S比色法硝酸-高氯酸消解后,在乙酸-乙酸钠缓冲液中,三价铝与铝-铬天青S及CTMAB反应生成蓝绿色络合物,于640nm波长测定吸光度铝总汞甲基汞面制食品中铝的测定苯芴酮比色法消解后,弱酸性溶液中,四价锡离子与苯芴酮生成微溶性橙红色络合物,在保护性胶体存在下,比色定量最低2mg/kg原子荧光光谱法消解后,锡被氧化成四价锡,用硼氢化钾或钠还原成硒化氢,原子荧光计中,生成锡原子,产生原子荧光。

食品安全国家标准食品理化检验方法总则范围本标准规定了食品理化检验方法的检验基本原则和要求。
本标准适用于食品安全标准检验方法理化部分。
术语与定义1.1 特异性:指方法定性区分待测物和其它物质的能力。
1.2 准确度:指检测结果与样品真值间的一致程度,准确度大小由定量的正确度和精密度决定。
1.3 精密度:指检测结果间的一致程度,通常用相对标准偏差表示。
1.4 重复性:指在同一实验室在人员、设备、方法等恒定条件下,在短时间内对同一测定对象进行独立测定的精密度。
1.5 再现性:指在不同实验室间,仅在方法相同的条件下对同一测定对象进行独立测定的精密度。
样品采集、保存与检验1.6 样品采集基本要求样品采集应有完整的采样信息如生产日期、批号、数量、生产者等,采集的样品应具有代表性和均匀性。
当样品量较大时需要采用四分法选出能反应该食品的卫生质量和满足检验项目样品量需要的检测样品,一式三份,供检验、复验、备查或仲载,一般每份样品不少于0.5kg,但掺伪食品和食物中毒样品除外。
1.7 样品包装建议有包装产品应采集包装产品,散装产品应根据所需开展的检验项目,采用适宜的、且可真实反映产品特性的容器。
1.8 液体、半流体食品植物油、鲜乳、酒或其他饮料和用大桶或大罐盛装的大包装产品应先充分混匀后再采样,并分层采样。
1.9 粮食及固体食品应自每批食品上、中、下三层中的不同部位分别采部分样品,采样量应符合相关标准要求,混合后按四分法对角取样,再进行几次混合,最后取有代表性样品。
1.10 肉类、水产等食品应按分析项目要求分别采取不同部位的样品或混合后采样。
1.11 罐头、瓶装食品或其他小包装食品应根据批号随机取样,同一批号取样件数,250g 以上的包装不得少于6个,250g 以下的包装不得少于10个。
1.12 掺伪食品和食物中毒的样品采集掺伪食品和食物中毒的样品要尽可能反映出其可能具有的中毒因素。
1.13 样品保存定型包装产品应在产品规定有效期按样品的保存条件予以保存,散装产品应参照相关产品的保存条件予以保存,且应采取有效措施保证样品不变质。


食品理化检验食品中重金属及有害元素的测定一、食品中铅的测定及限量标准(一)基本方法及方法要点:本标准检出限:石墨炉原子吸收光谱法为5µg/kg;氢化物原子荧光光谱法固体样品为5µg/kg,液体样品为1µg/kg;火焰原子吸收光谱法为0.1mg/kg;比色法为0.25 mg/kg.单扫描极谱法检出限为0.17µg。
(二)石墨炉原子吸收光谱法1.样品预处理:粮食、豆类去杂物后,磨碎、过20目筛、储于塑料瓶中、保存备用。
蔬菜、水果、鱼类、肉类及蛋类等水分含量高的鲜样,用食品加工机或匀浆机打成匀浆,储于塑料瓶中,保存备用。
样品消解可采用压力消解罐消解法、干法灰化、对硫酸铵灰化法和湿式消解法。
(见GB/T5009.12-2003)。
2.测定:基体改进剂的使用:对有干扰样品,注入适量的基体改进剂磷酸二氢铵(20g/L)一般为5µl或与样品同量消除干扰。
绘制铅标准曲线时也要加入与样品测定时等量的基体改进剂磷酸二氢铵溶液。
(三)火焰原子吸收光谱法:1.样品预处理:萃取法分离法:视样品情况,吸取25~50ml上述制备的样液及试剂空白液,分别置于125ml分液漏斗中补加水至60ml。
加2ml柠檬酸铵溶液,溴百里酚蓝指示剂3~5滴,用氨水(1:1)调PH至溶液有黄变蓝,加硫酸铵溶液10ml,DDTC溶液10ml,摇匀。
放置5分钟左右,加入10.0mlMIBK,剧烈振摇提取1分钟,静置且分层后,弃去水层,将MIBK层放入10ml带塞刻度管中备用。
2.测定:饮品、酒类及包装材料浸泡液可经萃取直接进样测定。
萃取液进样磨口适当减小乙炔气的流量。
(四)双硫腙比色法(五)食品中铅限量指标二、食品中镉的测定及限量标准(一)基本原理及方法要点:最低检出浓度:石墨炉原子化法为0.1µg/kg;火焰原子化法为5.0µg/kg;比色法为50µg/kg;标准曲线线性范围为0~50n g/ml。

水分测量(直接干燥法)1、该方法,设备操作简单,但时间较长,且不适合胶体、高脂肪、高糖食品及含有较多高湿、易氧化、易挥发物质的食品。
2、该方法测的水分还包括微量的芳香油、醇,有机酸等挥发性物质。
3、加入海沙是为了增加受热与蒸发面积,防止食品结块,加速水分蒸发,缩短分析时间。
4、水分蒸发干净与否,无直观指标,只能依靠衡重来判断。
衡重指的是两次烘烤称量的质量差不超过规定的毫克数,一般不超过2mg。
5、精密度:在重复条件下获得两次独立测定结果的绝对差值不得超过算术平均值的5%。
本方法最低检出量为0.002g,取样量为2.0mg时,该法检出限为0.1000g/100g.蛋白质的测量(凯氏定氮法)1、该方法精密度为10%,适用于各类食品中蛋白质的测定,但不适合用于添加无机含氮物质或有机非蛋白质含氮蒸物质的食品测定。
2、消化过程中应注意不时转动凯氏烧瓶,以便利用冷凝酸液将附在瓶壁上的固体残渣洗下,并促进消化完全。
3、蒸馏过程中,注意检查不能使系统漏气。
4、蒸馏℃时,蒸汽发生要均匀、充足。
加碱要够量,动作要快,防止氨损失。
5、冷凝管出口应浸入吸收液中,防止氨损失,若吸收液面过低,可适量补充少量水分,以确保冷凝管出口浸入吸收液中。
6、蒸馏过程中,宜始终保持蒸汽发生器中的水呈沸腾状态,以节约蒸馏时间,防止倒吸。
7、硼酸吸收液的温度不应超过40℃,否则对氨的吸收作用减弱而造成损失,此时可至于冷水浴中使用。
8、混合指示剂在碱性溶液中呈绿色,在中性溶液中呈灰色,在酸性条件下呈红色。
脂肪的测定(索氏提取法)1、本法为乳及乳制品脂肪含量测定的国际标准方法,适用于各种液态乳、各种炼乳、乳粉、奶油、冰激淋等能在碱性溶液中溶解的乳制品。
2、乳类脂肪虽然属于游离态脂肪,但因脂肪球被酪蛋白钙盐包裹,又处于高度分散的胶体体系中,故不能直接被乙醚、石油醚提取,需要先用氨水处理,使酪蛋白盐成为可溶性盐,所以该法又叫碱性乙醚抽提法。
3、加氨水后,要充分混匀,否则会影响下一步迷对脂肪的提取。


名词解释:1.恒量:是指在规定的条件下,连续两次干燥或灼烧后称定的质量差异不超过规定的范围。
2.空白试验:指除不加样品外,采用完全相同的分析步骤、试剂和用量(滴定法中标准滴定液的用量除外),进行平行操作所得的结果。
用于扣除样品中试剂本底和计算检验方法的检出限。
3.准确称取:指用精密天平进行的称量操作,其精度为±0.0001g。
4.农药残留:指农药使用后残存于生物体、食品(农副产品)和环境中的微量农药原体、有毒代谢物、降解物和杂质的总称,具有毒理学意义。
残存的数量称为残留量。
5.兽药残留:是指动物性产品中含有某种兽药的原形或其代谢物以及与兽药有关的杂质的残留。
6.粗蛋白:蛋白质的测定通常采用半微量凯氏定氮法和自动定氮分析法,这两种方法测得的均为食品中的总氮量(蛋白氮+非蛋白氮),故称为粗蛋白。
7.粗脂肪:样品用无水乙醚或石油醚等溶剂抽提后,蒸去溶剂所得的物质,在食品分析上称为脂肪或粗脂肪。
8采样:从一批食品中选出某一特定部分、一定数量的包装产品或单位产品,所取部分(样品)对被取样的整批食品最具代表性,供分析检验用。
第一章1、食品理化检验常用方法可分四大类:感官检查、物理检测、化学分析法、仪器分析法。
2、感官检查是指利用人体的感觉器官(眼、耳、鼻、口、手等)的感觉,即视觉、听觉、嗅觉、味觉和触觉等对食品的色、香、味、形和质等进行综合性评价的一种检验方法。
如果食品的感官检查不合格,或者已经发生明显的腐败变质,则不必再进行营养成分和有害成分的检测,直接判断为不合格食品。
一般嗅觉的敏感度远高于味觉。
触觉检查:用手接触食品,检查食品的轻重、软硬、弹性、粘稠、滑腻等性质。
对于鱼、肉制品、海产品等应检查食品的组织状态、新鲜程度、保存效果等现象。
2、相对密度:测液体浓度与纯度相对密度的测定:液态食品的相对密度可反映液态食品的浓度和纯度。
测定液体食品的相对密度可初步判断该食品质量是否正常。
脱脂乳的相对密度比全牛乳高;掺水牛乳相对密度降低。
《食品理化检验》课程标准1.课程简介食品加工技术课程是我院食品加工专业的专业核心课程,课程以工学结合为切入点。
本课程以“培养学生理解现代的食品加工技术的基本理论、基本知识,掌握典型食品的生产方法和工艺技术等基本技能,并具备良好的职业素养”为教学目标,使学生掌握最基本的食品加工的工艺知识,为今后进一步学习食品领域的专业课程或从事食品科研、产品开发、工业生产管理及相关领域的工作打下基础。
该课程与职业资格证书的考核要求紧密,通过该课程的学习,学生参加职业技能鉴定,获取中级职业资格证书,培养学生的职业能力。
该课程教学团队紧密结合企业发展需求实际,以就业为导向、以职业能力培养为重点、以工作过程为依据、以实训项目为载体、以职业技能和职业素养的培养为主旨,充分体现职业性、实践性和开放性的要求,更好地激发了学生学习兴趣,方便了因材施教。
本课程与职业资格证书的关系食品加工技术是一门具有较强的实践性、理论性的学科,是食品加工专业的主干课程和核心课程。
课程发展的早期阶段:本课程以各种食品的生产工艺为主线,通过食品加工环节和过程展示,培养学生理论联系实际,解决生产中的实际问题。
课程发展的改革阶段:随着市场经济的发展和人才的需求结构的变化,课程小组成员从理论和实践实训两方面进行教学改革。
对课程内容进一步精选,整合和精简,重构少而精的教学内容。
理论够用就行,加大实践教学内容。
按照项目教学法,从培养学生的动手能力出发,整个教学内容分为8个项目。
同时,在课堂内容上强调基本原理及共性部分的讲授,通过代表性的实验帮助学生掌握有关教学内容。
完善了现有教材的配套相关教学资源。
3.课程在人才培养方案中的地位和作用核心课程:对食品生产加工能力进行培养的一门专业学习领域课程,将人才培养目标定为培养熟练掌握食品加工及管理基本知识与技能,具备一定的产品创新及研发能力的高技能人才。
前导课程:有机化学、无机化学、食品生物化学、食品保藏等;支撑后续课程:食品质量管理、食品安全与卫生、食品综合实训、顶岗实习等。
食品安全生产(理化)检测国家标准内容祥解
本文选择了食品检验常用的理化标准,对标准号、适用范围与要点进行了梳理,期望对食品检验领域的小伙伴们的学习提供帮助。
一、食品中水分的测定
二、食品中灰分的测定
三、食品中蛋白质的测定
注:当只检测氮含量时,不需要乘蛋白质换算系数F 四、食品中脂肪的测定
⑧滴定过程快速避光;取样2-3g;平均值结果保留2位有效数字。
计算公式:
①试样称样量和滴定液浓度应使滴定液用量在0.2mL~10mL之间(扣除空白后)。
若
检测后,发现样品的实际称样量与该样品酸价所对应的应有称样量不符,应按照表1要求,调整称样量后重新检测。
②酚酞指示剂,当试样溶液初现微红色,15s内无明显褪色时,为滴定终点。
平均值结果,除另有规定外,有效数字小数点后两位。
截止2022年2月,食品安全国家标准共计1419项,每个标准都有不同的适用范围,有些品种还不只一个相关标准。
4月又有一批新标准要开始实施了。
在这里给大家一个建议,打开国标第一步不要盲目就开始实验,一定要先看适用范围选择合适的方法,再看检验方法对不同品种在实验条件上的区别,选择对的实验条件,谋定而后动,才能少走弯路哦~。
食品理化检验标准汇编食品理化检验标准众多,其中有较为通用的也有与具体产品相关联的。
食品580收集整理了通用的理化检验标准供大家参考使用。
1GB类GBT 10247-2008 粘度测量方法GBT 12456-2008 食品中总酸的测定GBT 12457-2008 食品中氯化钠的测定GBT 14772-2008 食品中粗脂肪的测定GBT 18979-2003 食品中黄曲霉毒素的测定免疫亲和层析净化高效液相色谱法和荧光光度法GBT 21911-2008 食品中邻苯二甲酸酯的测定GBT 21915-2008 食品中纳他霉素的测定液相色谱法GBT 21927-2008 食品中叔丁基对苯二酚的测定高效液相色谱法GBT 22110-2008 食品中反式脂肪酸的测定气相色谱法GBT 22220-2008 食品中胆固醇的测定高效液相色谱法GBT 22221-2008 食品中果糖葡萄糖蔗糖麦芽糖乳糖的测定高效液相色谱法GBT 22223-2008 食品中总脂肪、饱和脂肪(酸)、不饱和脂肪(酸)的测定水解提取-气相色谱法GBT 22224-2008 食品中膳食纤维的测定酶重量法和酶重量法-液相色谱法GBT 22235-2008 液体粘度的测定GBT 22253-2008 食品中阿力甜的测定GBT 22254-2008 食品中阿斯巴甜的测定GBT 23372-2009 食品中无机砷的测定液相色谱电感耦合等离子体质谱法GBT 23373-2009 食品中抗氧化剂丁基羟基茴香醚(BHA)、二丁基羟基甲苯(BHT)与特丁基对苯二酚(TBHQ)的测定GBT 23374-2009 食品中铝的测定电感耦合等离子体质谱法GBT 23377-2009 食品中脱氢乙酸的测定高效液相色谱法GBT 23378-2009 食品中纽甜的测定方法高效液相色谱法GBT 23381-2009 食品中6-苄基腺嘌呤的测定高效液相色谱法GBT 23382-2009 食品中丙酸钠、丙酸钙的测定高效液相色谱法GBT 23383-2009 食品中双乙酸钠的测定高效液相色谱法GBT 23490-2009 食品水分活度的测定GBT 23495-2009 食品中苯甲酸、山梨酸和糖精钠的测定高效液相色谱法GBT 23496-2009 食品中禁用物质的检测碱性橙染料高效液相色谱法GBT 23499-2009 食品中残留过氧化氢的测定方法GBT 23501-2009 食品中T-2毒素的测定免疫亲和层析净化高效液相色谱法GBT 23502-2009 食品中赭曲霉毒素A的测定免疫亲和层析净化高效液相色谱法GBT 23503-2009 食品中脱氧雪腐镰刀菌烯醇的测定免疫亲和层析净化高效液相色谱法GBT 23504-2009 食品中玉米赤霉烯酮的测定免疫亲和层析净化高效液相色谱法GBT 23749-2009 食品中叶绿素铜钠的测定分光光度法GBT 23813-2009 食品中1,2-丙二醇的测定GBT 5009.11-2003 食品中总砷及无机砷的测定GBT 5009.1-2003 食品卫生检验方法理化部分总则GBT 5009.120-2003 食品中丙酸钠、丙酸钙的测定GBT 5009.121-2003 食品中脱氢乙酸的测定GBT 5009.124-2003 食品中氨基酸的测定GBT 5009.128-2003 食品中胆固醇的测定GBT 5009.13-2003 食品中铜的测定GBT 5009.132-2003 食品中莠去津残留量的测定GBT 5009.137-2003 食品中锑的测定GBT 5009.138-2003 食品中镍的测定GBT 5009.141-2003 食品中诱惑红的测定GBT 5009.14-2003 食品中锌的测定GBT 5009.149-2003 食品中栀子黄的测定GBT 5009.150-2003 食品中红曲色素的测定GBT 5009.151-2003 食品中锗的测定GBT 5009.154-2003 食品中维生素B6的测定GBT 5009.157-2003 食品中有机酸的测定GBT 5009.159-2003 食品中还原型抗坏血酸的测定GBT 5009.168-2003 食品中二十碳五烯酸和二十二碳六烯酸的测定GBT 5009.169-2003 食品中牛磺酸的测定GBT 5009.17-2003 食品中总汞及有机汞的测定GBT 5009.18-2003 食品中氟的测定GBT 5009.191-2006 食品中氯丙醇含量的测定GBT 5009.19-2008 食品中有机氯农药多组分残留量的测定GBT 5009.20-2003 食品中有机磷农药残留量的测定GBT 5009.208-2008 食品中生物胺含量的测定GBT 5009.210-2008 食品中泛酸的测定GBT 5009.215-2008 食品中有机锡含量的测定GBT 5009.2-2003 食品的相对密度的测定GBT 5009.22-2003 食品中黄曲霉毒素B1的测定GBT 5009.23-2006 食品中黄曲霉毒素B1、B2、G1、G2的测定GBT 5009.26-2003 食品中N-亚硝胺类的测定GBT 5009.27-2003 食品中苯并(a)芘的测定GBT 5009.29-2003 食品中山梨酸、苯甲酸的测定GBT 5009.30-2003 食品中叔丁基羟基茴香醚(BHA)与2,6-二叔丁基对甲酚(BHT)的测定GBT 5009.31-2003 食品中对羟基苯甲酸酯类的测定GBT 5009.34-2003 食品中亚硫酸盐的测定GBT 5009.35-2003 食品中合成着色剂的测定GBT 5009.6-2003 食品中脂肪的测定GBT 5009.7-2008 食品中还原糖的测定GBT 5009.8-2008 食品中蔗糖的测定GBT 5009.82-2003 食品中维生素A和维生素E的测定GBT 5009.83-2003 食品中胡萝卜素的测定GBT 5009.84-2003 食品中硫胺素(维生素B1)的测定GBT 5009.85-2003 食品中核黄素的测定GBT 5009.87-2003 食品中磷的测定GBT 5009.89-2003 食品中烟酸的测定GBT 5009.90-2003 食品中铁、镁、锰的测定GBT 5009.91-2003 食品中钾、钠的测定GBT 5009.9-2008 食品中淀粉的测定GBT 5009.97-2003 食品中环己基氨基磺酸钠的测定2SB类SBT 10317-1999 蛋白酶活力测定法SBT 10318-1999 氨态氮测定法SBT 10319-1999 熟料消化率测定法SBT 10320-1999 熟料N型蛋白试验SBT 10321-1999 水溶性物的样品制备SBT 10322-1999 pH测定法SBT 10323-1999 色度测定法SBT 10326-1999 无盐固形物测定法3SN类SN 0180-1992 食品水活度测定方法SNT 0448-2011 进出口食品中砷、汞、铅、镉的检测方法电感耦合等离子体质谱(ICP-MS)法SNT 0864-2011 出口食品中铝的测定电感耦合等离子体质谱法SNT 1547-2011 进出口食品中甲醛的测定液相色谱法SNT 1945-2007 食品中反式脂肪酸含量的测定方法毛细管气相色谱法SNT 2115-2008 进出口食品和饲料中总氮和粗蛋白的检测方法杜马斯燃烧法SNT 2326-2009 食品及油脂中反式脂肪酸含量的检测傅立叶变换红外光谱法SNT 2918-2011 出口食品中亚硫酸盐的检测方法离子色谱法SNT 2922-2011 出口食品中EPA和DHA的测定气相色谱法SNT 3032-2011 出口食品中三聚氰胺和三聚氰酸检测方法液相色谱质谱质谱法SNT 3142-2012 出口食品中D-甘露糖醇、麦芽糖、木糖醇、D-山梨糖醇的检测方法液相色谱质谱质谱法SNT 3147-2012 出口食品中邻苯二甲酸酯的测定SNT 3148-2012 出口食品中过氧化苯甲酰含量的测定高效液相色谱法SNT 3149-2012 出口食品中三苯锡、苯丁锡残留量检测方法气相色谱质谱法SNT 3151-2012 出口食品中亚硝酸盐和硝酸盐的测定离子色谱法SNT 3543-2013 出口食品中六溴环十二烷的测定液相色谱质谱质谱法SNT 3544-2013 出口食品中全氟辛酸和全氟辛烷磺酸盐的测定液相色谱质谱质谱法SNT 3727-2013 进出口食品中碘含量的测定离子色谱法4QB、NY类NYT 1723-2009 食品中富马酸二甲酯的测定高效液相色谱法QBT 2186-1995 氨气敏电极法测定水解蛋白液含氮量。
食品安全国家标准食品理化检验方法总则范围本标准规定了食品理化检验方法的检验基本原则和要求。
本标准适用于食品安全标准检验方法理化部分。
术语与定义1.1 特异性:指方法定性区分待测物和其它物质的能力。
1.2 准确度:指检测结果与样品真值间的一致程度,准确度大小由定量的正确度和精密度决定。
1.3 精密度:指检测结果间的一致程度,通常用相对标准偏差表示。
1.4 重复性:指在同一实验室在人员、设备、方法等恒定条件下,在短时间内对同一测定对象进行独立测定的精密度。
1.5 再现性:指在不同实验室间,仅在方法相同的条件下对同一测定对象进行独立测定的精密度。
样品采集、保存与检验1.6 样品采集基本要求样品采集应有完整的采样信息如生产日期、批号、数量、生产者等,采集的样品应具有代表性和均匀性。
当样品量较大时需要采用四分法选出能反应该食品的卫生质量和满足检验项目样品量需要的检测样品,一式三份,供检验、复验、备查或仲载,一般每份样品不少于0.5kg,但掺伪食品和食物中毒样品除外。
1.7 样品包装建议有包装产品应采集包装产品,散装产品应根据所需开展的检验项目,采用适宜的、且可真实反映产品特性的容器。
1.8 液体、半流体食品植物油、鲜乳、酒或其他饮料和用大桶或大罐盛装的大包装产品应先充分混匀后再采样,并分层采样。
1.9 粮食及固体食品应自每批食品上、中、下三层中的不同部位分别采部分样品,采样量应符合相关标准要求,混合后按四分法对角取样,再进行几次混合,最后取有代表性样品。
1.10 肉类、水产等食品应按分析项目要求分别采取不同部位的样品或混合后采样。
1.11 罐头、瓶装食品或其他小包装食品应根据批号随机取样,同一批号取样件数,250g 以上的包装不得少于6个,250g 以下的包装不得少于10个。
1.12 掺伪食品和食物中毒的样品采集掺伪食品和食物中毒的样品要尽可能反映出其可能具有的中毒因素。
1.13 样品保存定型包装产品应在产品规定有效期按样品的保存条件予以保存,散装产品应参照相关产品的保存条件予以保存,且应采取有效措施保证样品不变质。