ChemOffice2008实用教程
- 格式:ppt
- 大小:9.98 MB
- 文档页数:66


ChemDraw的简单使用教程1、ChemDraw的简介ChemDraw软件是目前国内外最流行、最受欢迎的化学绘图软件。
它是美国CambridgeSoft公司开发的ChemOffice系列软件中最重要的一员。
由于它内嵌了许多国际权威期刊的文件格式,近几年来成为了化学界出版物、稿件、报告、CAI软件等领域绘制结构图的标准。
ChemDraw是世界上使用最多的大型软件包ChemOffice中的一个组件,其它两个组件为Chem3D(分子结构模型)和ChemFinder(化学数据库信息),其最新版本是Chem Office 2004(ChemDraw Pro 8.0、Chem3D Ultra8.0、ChemFinderUltra8.0) 。
ChemDraw可以建立和编辑与化学有关的一切图形。
例如,建立和编辑各类化学式、方程式、结构式、立体图形、对称图形、轨道等,并能对图形进行翻转、旋转、缩放、存储、复制、粘贴等多种操作。
基于国际互联网技术开发的智能型数据管理系统,包含的多种化学通用数据库共四十多万个化合物的性质、结构、反应式、文献等检索条目的分析和利用,可为化学家的目标化合物设计、反应路线选择和物化性质预测以及文献的调用提供极大的方便。
该软件可以运行于Windows平台下,使得其资料可方便地共享于各软件之间。
除了以上所述的一般功能外,其ultra版本还可以预测分子的常见物理化学性质如:熔点、生成热等;对结构按IUPAC原则命名;预测质子及碳13化学位移动等。
2、ChemDraw的基础知识1、菜单栏:含有操作chemdraw应用文件和内容的命令设置。
2、工具栏:含有常用命令图标,单击图标时,效果与选择菜单中相应的命令一样。
3、编辑区:供绘制图形结构的工作区。
4、滚动栏:含有滚动框、滚动按钮和滚动条。
5、状态栏:标出当前的工作内容以及鼠标指到某些菜单按钮时的说明图形工具板含有所有能够在文件窗口绘制结构图形的工具,选择了这些工具的图标后,光标将随之改变成为相应的工具形状。

C hemoffice2008使用教程ChembioOffice简介ChembioOffice是由CambridgeSoft开发的综合性科学应用软件包。
该软件包是为广大从事化学、生物研究领域的科研人员个人使用而设计开发的产品。
同时,这个产品又可以共享解决方案,给研究机构的所有科技工作者带来效益。
利用ChemBioOffice你可以方便的进行化学生物结构绘图、分子模型及仿真、可以将化合物名称直接转为结构图,省去绘图的麻烦;也可以对已知结构的化合物命名,给出正确的化合物名。
目前最新的版本为Chemoffice 2010。
Chemoffice2008包括以下几个部分:1.ChemDraw Ultra11.0:化学结构绘图。
2.Chem3D Ultra11.0:分子模型及仿真。
3.ChemFinder Ultra11.0:化学信息搜寻整合系统4.E-Notebook11.0:整理化学信息、文件和数据,并从中取得所要的结果。
Chemoffice安装方法:1.运行setup.exe2.输入证书时输入:Name:lZ0email:lz0@Serial:011-256104-71993.然后点击"Activate Later"4.复制crack.reg文件到C:\Program Files\CambridgeSoft\ChemOffice2008\Common\DLLs 双击确定即可ChemDraw的使用方法:1.绘制结构式以水杨酸(salicylic acid)为例步骤:1.打开ChemDraw2.单击垂直工具栏右下角的图标,鼠标变成苯环的样子,在绘图区单击鼠标出现一个苯环3.单击图标,将鼠标移至苯环的一个角上,出现深色的正方形连接点4.自连接点横向拉出一根实线单键,松开鼠标,自单键终点向右下方再次拉出一根单键,与前一根单键夹角约为109°,其余类推。
5.选中垂直工具栏中的文本工具,分别将鼠标移至应该出现羟基或氧原子的位置,待出现连接点后,点击键盘上的O键。





绝对实⽤chemdraw教程教案资料绝对实⽤c h e m d r a w教程ChemDraw软件Chemdraw7.0是Cambridgesoft公司出品的Chemoffice系列的最新版本Chemoffice2002的重要组成部分。
⼀、ChemDraw软件简介1、启动:在Windows下,有两种打开⽅式:在桌⾯上,双击ChemDraw图标;或者在“开始菜单”⾥选择ChemDraw选项。
2、界⾯简介:下图为ChemDraw的图形化⽤户界⾯(GUI)。
关于画图的主要⼯具——⼯具栏,其主要功能如下图所⽰:仅供学习与交流,如有侵权请联系⽹站删除谢谢2ChemDraw为绘制化学结构,提供了以下⼯具。
化学键⼯具, 环⼯具, 脂肪链⼯具, ⾃动错误矫正, 在ChemDraw Ultra 中还新增了从化学名称到结构的功能。
⑴、化学键的绘制:绘制化学键有以下⼏种⼯具,利⽤这些⼯具,可以直接在⽂档中单击或者拖拉来绘制化学键。
默认的键长和键⾓⽤Object菜单中Fixed Lengths 和Fixed Angles选项固定。
仅供学习与交流,如有侵权请联系⽹站删除谢谢3仅供学习与交流,如有侵权请联系⽹站删除谢谢4⑵、环的绘制:环的绘制,可以使⽤右边的⼯具:环的绘制可以⽤以下的⽅法,注意以下的细节:如果选择环⼯具之后,在⼀个原⼦上单击,则会在该原⼦上添加⼀个环,如果在⼀条键上单击,则会在键上添加环,具体的演⽰如下:如果想要绘制共轭环结构,可以按下CTRL 键的同时,在⽂档中单击放置环。
这样,会在环中出现⼀个圆圈。
当然,这种情况下,环已烷的椅式结构除外。
⑶、脂肪链键的绘制:选择脂肪链⼯具,在⽂档上拖动⿏标即可画出长链。
也可以在⽂档空⽩区域或者⼀个原⼦上单击,在出现的对话框输⼊想要链长的原⼦数。
⑷、键的编辑:改变键的类型:选择键⼯具,指向已经存在的键,单击,则会出现新的类型的键。
改变楔形键和虚格键的⽅向:选择楔形键和虚格键⼯具,点击适当的位置,则⽅向被⾃动调整。
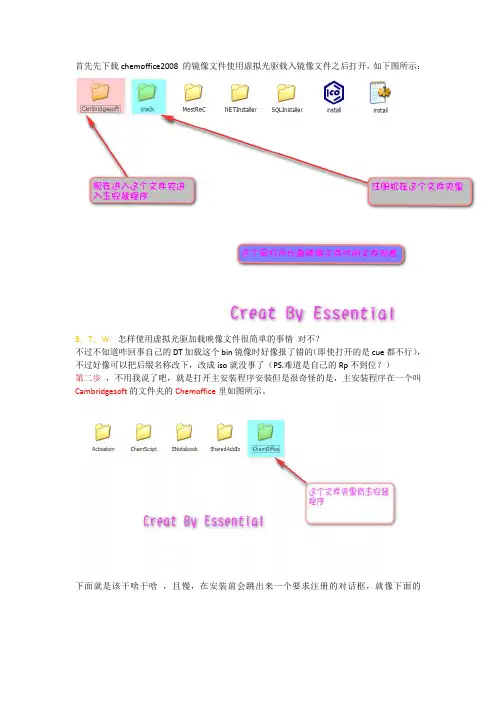
首先先下载chemoffice2008 的镜像文件使用虚拟光驱载入镜像文件之后打开,如下图所示:
B.T.W.怎样使用虚拟光驱加载映像文件很简单的事情对不?
不过不知道咋回事自己的DT加载这个bin镜像时好像报了错的(即使打开的是cue都不行),不过好像可以把后缀名称改下,改成iso就没事了(PS.难道是自己的Rp不到位?)
第二步,不用我说了吧,就是打开主安装程序安装但是很奇怪的是,主安装程序在一个叫Cambridgesoft的文件夹的Chemoffice里如图所示。
下面就是该干啥干啥,且慢,在安装前会跳出来一个要求注册的对话框,就像下面的
现在打开注册机(呵呵,注册机在那里?大家看第一幅图的说明)就像是这样
现在就按找第三幅的提示办~!Creat By Essential。

计算化学实验Chem3D软件指南Chem3D开始:点击桌面“Chem3D Ultra 10.0”图标Chem3D一览菜单和工具栏区组成区坐标区光谱区输出结果区构象区2D分子区3D绘图区Chem3D详解菜单和工具栏区文件打开输入打印设置编辑拷贝粘贴修改选择显示工具结构测量旋转饱和修正重叠计算界面结果密度面轨道电势面分子面电影旋转轨迹数据库窗口排列帮助说明例子Chem3D详解文件新分子模型样本分子打开文件中的分子结构保存Chem3D详解编辑撤消操作重复操作清除当前分子Chem3D详解视图分子显示方式显示取向工具栏分子组成ChemDraw直角坐标Z-矩阵测量列表参数表输出区构象区光谱显示区文件输入原子类型MM2类型Chem3D详解结构测量列表分子位置和取向设置Z-matrix饱和价键(加氢)自动调整分子结构重叠分子对接Chem3D详解计算模块计算结果管理停止计算二面角计算(构象搜索)EHMO计算电荷轨道MM2分子动力学模块分子性质计算模块GAMESS计算界面Gaussian计算界面Jaguar计算界面Mopac计算界面Chem3D详解计算模块二面角计算(构象搜索)扫描一个二面角扫描两个二面角二面角计算(构象搜索) 扫描一个二面角二面角计算(构象搜索) 扫描二个二面角构象过渡态Chem3D详解计算模块EHMO电荷分子面总电子密度面分子轨道Chem3D详解计算模块EHMO电荷Chem3D详解计算模块EHMO总电子密度面等值面图isoval=0.01Chem3D详解计算模块EHMO分子轨道等值面图isoval=0.01MM2分子动力学模块Chem3D详解计算模块运行MM2输入文件优化构型(能量极小化)分子动力学性质计算打印力场参数MM2分子动力学模块Chem3D详解计算模块力场参数M2 Constant Value QualityCubic stretch constant-2.000 4Quarticstretch constant2.333 4X-B,C,N,O-Y Stretch-Bend interaction force constant0.120 4 X-B,C,N,O-H Stretch-Bend interaction force constant0.090 4 Sexticbending constant (* 10**8)7.000 4Cutoff distance for charge/charge interactions35.000 4Cutoff distance for charge/dipole interactions25.000 4Cutoff distance for dipole/dipole interactions18.000 4Cutoff distance for vander Waalsinteractions10.000 4MM2 Atom RadiusEps WeightReduct Lone Pairs Quality 5 1.500 0.047 1.008 0.915 0 4 2 1.940 0.044 12.000 0.000 0 4 Bond KS Bond Length Dipole Quality2-5 4.600 1.100 0.000 42-2 9.600 1.337 0.000 4Angle KB XR2 XRH XH2 Quality 2-2-5 0.360 120.0 120.5 0.0 42-2-2 0.430 120.0 0.0 0.0 4Atoms Force Constant Quality2-2 0.050 42-5 0.050 4Torsional V1 V2 V3 Quality5-2-2-5 0.000 15.000 0.000 42-2-2-5 0.000 9.000 -1.060 42-2-2-2 -0.930 8.000 0.000 4PiAtom Electron Ionization Repulsion Quality 2 1 -11.160 11.134 4PiBond DForce DLength Quality2-2 4.600 0.166 43, 4次键伸缩常数键伸缩-弯曲作用参数截断距离LJ非键参数键伸缩参数键弯曲(角)参数键扭转参数π键(共轭)修正参数MM2分子动力学模块Chem3D详解计算模块优化构型(能量极小化)收敛要求: 最小均方根梯度(力)输出结果能量分解总能量MM2分子动力学模块Chem3D详解计算模块分子动力学模拟步长结构输出间隔轨迹步数升温速率目标温度参数一览MM2分子动力学模块Chem3D详解计算模块分子动力学模拟暂停/开始停止MM2分子动力学模块Chem3D详解计算模块分子动力学模拟输出结果时间(10-15秒)总能势能温度MM2分子动力学模块Chem3D详解计算模块分子动力学模拟输出结果分子平动能= 0分子转动能= 0非平衡平衡态MM2分子动力学模块Chem3D详解计算模块分子动力学模拟输出结果总能守恒!温度控制!MM2分子动力学模块Chem3D详解计算模块π键键级能量Chem3D详解计算模块分子性质计算模块Chem3D详解计算模块GAMESS优化构型优化过渡态分子性质频率分析红外光谱NMR光谱创建输入文件查看输出结果文件运行输入文件Chem3D详解计算模块GAMESS作业类型方法基组波函数类型基组极化函数基组扩散函数基组指数优化方法限制性部分优化坐标自旋多重度= 单电子数+1净电荷Chem3D详解计算模块GAMESS高级设置-1最大SCF循环次数优化最大步数收敛要求(力)温度(仅用于热力学计算)溶剂模型溶剂参数初始猜测方法分子对称性点群内存要求Chem3D详解计算模块GAMESS性质偶极电子密度静电势Lowdin电荷和布居分析Mulliken电荷和布居分析电子势能电子总能量电子动能Chem3D详解计算模块GAMESS计算方法激发态从头算理论密度泛函理论基态Chem3D详解计算模块GAMESS等电子密度面(0.001)HF/STO-3G IR光谱苯甲醇NMR光谱Mopac计算界面Chem3D详解计算模块优化平衡构型优化过渡态分子性质IR光谱Mopac计算界面Chem3D详解计算模块作业类型方法波函数优化方法溶剂模型限制性优化收敛要求坐标Mopac计算界面Chem3D详解计算模块电荷偶极静电势生成焓超精细耦合常数电离能电荷溶剂介电常数Chem3D详解分子面模块分子面探测半径显示模式颜色模式分辨率选择分子轨道等值面值溶剂面总电子密度面总自旋密度面静电势面分子轨道清除所有面Chem3D详解分子面模块SA面总电子密度面isoval=0.01总电子密度面/静电势isoval=0.01静电势isoval=1Connolly面HOMO轨道LUMO轨道甲醇Chem3D详解工具栏新分子打开保存拷贝剪切粘贴恢复重复印模型背景特效红蓝镜像坐标原子符号原子编号残基全屏电影播放选择平移旋转缩放移动选中键型清除饱和调整符输入擦写分子面探测半径显示配色分辨率分子轨道等值状态优化旋转停止Chem3D详解文件格式晶体结构文件Gaussian输入输出文件cif文件pdb文件(蛋白质)Chem3D详解晶体结构文件有机分子晶体: CCDC数据库, 草酸晶体Pbca空间群(61)OXALAC06.datChem3D详解晶体结构文件无机晶体: ICSD, AMCSDAmerican Mineralogist Crystal Structural Database http://rruff.geo.arizona. edu/AMS/amcsd.php不能正确处理周期性的体系!!Chem3D详解生物大分子pdb文件(蛋白质)RSCB PDB蛋白质晶体结构数据库/pdb/home/home.do1AAQChem3D详解振动模式用JMOL显示GAMESS的频率分析输出文件需要把Gamess输出文件修改一下才能用Jmol5.0正确读出!将这些含“REDUCED MASSES”的行全部删除! 然后保存退出此处只留一个空行!Chem3D详解振动模式用JMOL显示GAMESS的频率分析输出文件旋转分子缩放平移选择测量轨迹/频率Chem3D详解振动模式用JMOL显示GAMESS的频率分析输出文件打开修改后的GAMESS输出文件*.out打开“extras”→“vibrate ...”Chem3D详解振动模式用JMOL显示GAMESS的频率分析输出文件选择频率观察“绕CO键的内转动”。
化学绘图类软件ChemWindow的使⽤⽅法和技巧Chem windows软件是化学绘图类软件,主要⽤于分⼦结构式的绘制。
ChemWindow 的操作绝⼤部分由⿏标便可完成,基本使⽤⽅法借助Help亦可较快学会。
化学绘图软件(ChemWindows) v6.0 汉化特别版(附序列号)类型:图像处理⼤⼩:14.3MB语⾔:简体中⽂时间:2014-02-27查看详情Chem Windows不但可以画⼆维分⼦结构,还提供了模拟三维分⼦结构的⼯具。
在中⽂Windows环境可直接输⼊汉字,所画分⼦结构式可通过剪贴,放⼊Word等for Windows软件编辑的⽂件中,还可利⽤其提供的程序转换为其它多种图形格式的⽂件,为其它应⽤软件调⽤,如转换为EPS⽂件。
ChemWindow 的使⽤⽅法和技巧:ChemWindow 的操作绝⼤部分由⿏标便可完成,基本使⽤⽅法借助Help亦可较快学会。
下⾯介绍作者在使⽤过程中的⼀些点滴经验和体会。
1.绘制ChemWindow 图形的⼏点技巧在绘制复杂分⼦结构式时,利⽤复制、⽔平或垂直翻转等操作可绘出对称协调的分⼦结构式图形。
⽽在绘制化学反应⽅程式时,当反应式很长时,各部分之间要处于同⼀⽔平线上,若采取逐个摆放,既费时⼜费⼒,最简单的⽅法是先⽤选择⼯具选取该反应式,然后从Arrange下拉菜单中选择Align Object命令,出现⼀对话框,选⽔平排序即可。
分⼦结构式中原⼦标记⽤标记⼯具(Label Tool)为好,虽然标题⼯具(Caption Tool)也能进⾏元素符号的输⼊,但⽤该⼯具输⼈的元素符号独⽴于所画的分⼦结构式,且元素符号与键之间连接很不协调,不便于后续操作。
⽽⽤标记⼯具输⼊,则元素符号与所画分⼦结构式构成⼀个协调的整体。
2. Word⽂本中ChemWindow 图形的嵌⼈和幻灯⽚的制作ChemWindow 图形以***.cw2形式存储,⼀般软件不能直接调⽤。
这样Word⽂本中嵌⼊分⼦结构式图形,⼀般遵循下列操作程序:开机→运⾏Windows→打开Word⽂本⽂件→最⼩化(切换⾄桌⾯)→运⾏ChemWindow→打开图形⽂件(或绘制分⼦结构式图形)→将所选图形复制到剪贴板→切换⾄Word⽂本中→将光标置于⽂本中需嵌⼈图形的位置,从编辑菜单中选择粘贴命令。
学生使用手册软件操作流程1.登陆与注册身份确认进入软件登陆画面,首先选择登陆身份,学生用户请选择“学生”注册第一次使用本软件的学生需要先进行注册,以便在系统中留下有关信息。
方法是点击[注册]按钮,弹出注册对话框,如图所示。
在对话框中输入学号等有关信息,其中“学号”、“密码”、“密码确认”是必填信息,其他为选填信息。
点击[确定],在确认无误后保存。
登陆已经注册的学生用户,在下次进入登陆画面的时候,可以直接输入学号和密码,并点击[登陆]按钮进入系统2.实验原始数据处理a)新增实验实验原始数据输入学生可以点击工具栏上新增实验按钮,或选择菜单【实验原始数据】→【新增实验】以清空“实验原始数据表”,然后即可在表上输入实验原始数据。
按钮[插入行]和[删除行]用于在输入时插入一个空白数据行和删除一个数据行。
登陆后首次进入主界面时,系统自动处于新增实验状态,可以直接输入实验数据。
实验保存实验数据输入完毕后必须进行保存:点击工具栏上实验保存按钮,或选择菜单【实验原始数据】→【保存实验】,弹出如图所示对话框。
从装置下拉式列表中选择实验所使用的装置,并点击[保存]按钮确定。
实验保存后就可以查看实验结果和曲线,具体操作见“实验结果显示与保存”说明。
b)编辑实验实验打开要对已有的实验进行编辑或查看实验结果和曲线需要先打开实验:点击工具栏上编辑实验按钮,或选择菜单【实验原始数据】→【编辑实验】,弹出如图所示对话框,从实验装置下拉列表中选择装置,然后在实验列表中选择所要打开的实验,点击[打开]按钮打开实验。
实验类型中,“基本型”表示用户手动输入数据的实验,“数字型”表示从MCGS导入数据的实验。
实验数据编辑修改实验打开后,可以直接在“实验原始数据表”上对实验数据进行修改。
要保存修改后的结果,请点击工具栏上实验保存按钮,或选择菜单【实验原始数据】→【保存实验】,确认之后保存。
另外,实验打开后可以对该实验的结果和曲线进行查看和保存,具体操作见“实验结果显示与保存”说明。