计算化学基础及ADF软件1
- 格式:pdf
- 大小:5.36 MB
- 文档页数:84
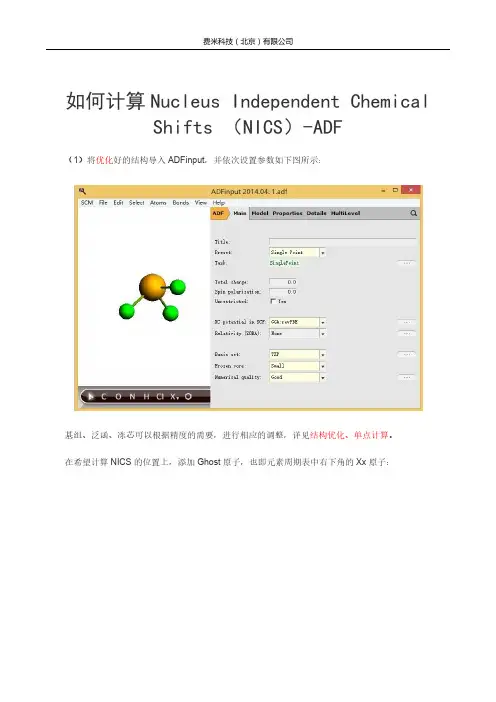
如何计算Nucleus Independent Chemical Shifts (NICS)-ADF
(1)将优化好的结构导入ADFinput,并依次设置参数如下图所示:
基组、泛函、冻芯可以根据精度的需要,进行相应的调整,详见结构优化、单点计算。
在希望计算NICS的位置上,添加Ghost原子,也即元素周期表中右下角的Xx原子:
在Properties—NMR菜单中将Ghost原子Xx选中,点击下图所示的“+”:
保存TAPE10文件:
取消对称性:
保存*.run文件。
(2)保存该文件后,运行该文件:
1)在Linux下:
●普通列表项目adfjobs—选中该任务—job—run
●ADFinput—File—Run
●在run文件所在目录直接执行./*run >*.out
2)在Windows下:
●adfjobs—选中该任务—job—run
●ADFinput—File—Run
●在run文件所在目录直接双击run文件执行,或右键点击run文件,然后选择“运行方式”中带adf
图标的方式,运行
(3)结果查看:
在生成的logfile文件末尾我们可以看到我们需要计算的两个点的化学位移屏蔽张量:
在out文件的末尾也可以看到类似更详细的内容:
(4)NICS:Ghost可以被看作是一个中子,因此该点的NICS的值为该点的屏蔽张量中的各向同性部分乘以-1——也就是logfile中,该点的NMR Shielding值乘以-1。

ADF教程如何去掉FCF计算中的虚频和低频模式ADF (Amsterdam Density Functional) 是一种常用的量子化学软件,用于计算分子结构、能量、振动频率等属性。
在进行FCF (Frequency Calculations) 计算时,有时候会出现虚频和低频模式,通常由于计算方法选择不当、初猜结构不合理或优化算法出现问题等原因引起。
本文将介绍如何去除FCF计算中的虚频和低频模式。
1.确认计算方法的准确性:首先要确保选择了适当的计算方法,如B3LYP、PBE等。
不同的计算方法对于不同分子体系可能会产生不同的计算结果。
因此,可以尝试使用不同的计算方法进行计算,并比较结果,以获得最准确的结果。
2.确认初猜结构的合理性:在进行FCF计算之前,通常需要对体系进行几何优化。
优化过程中,初始结构的选择是非常重要的。
不合理的初始结构可能导致优化结果不收敛或者出现虚频。
因此,在进行优化之前,可以使用其他方法获取一个合理的初始结构,例如根据实验结果得到的结构或者理论预测的结构。
3.调整优化相关参数:在进行几何优化计算时,ADF提供了一些相关的参数可以进行调整。
例如,可以调整优化的最大步长、收敛准则等参数。
适当调整这些参数可以提高优化的收敛速度和结果的准确性。
4. 使用更高级的计算级别:如果以上方法不能解决问题,可以尝试使用更高级的计算级别。
例如,可以尝试使用更高的基组函数,如6-311G(d,p)、cc-pVTZ等,以提高计算的精度。
同时,也可以尝试使用更高级的泛函,如M06、M11等。
5.检查输入文件和计算参数:有时候,虚频和低频模式的出现可能是由于输入文件或计算参数设置错误导致的。
因此,在进行计算之前,应该仔细检查输入文件和计算参数,确保没有错误。
特别是频率计算相关的参数,如计算的频率范围、计算的模式等。
6. 进行频率修正:如果上述方法无法解决问题,而且计算结果仍然包含虚频和低频模式,可以尝试进行频率修正。
ADF软件初学者使用指南一、安装运行ADF客户端软件1.用户下载ADF客户端软件,即ADF应用模型设置、前处理、结果分析软件。
2.下载地址:中国矿业大学主页之现代分析与计算中心网站,注册后,常用下载栏目之“ADF客户端软件之windows (32-bit)”3.在用户的PC机的windows操作系统中安装下载的ADF客户端软件:adf2012_windows.exe4.安装license许可:将license.txt文件放入ADF的安装目录(ADF2012版即adf2012.01目录中)。
5.运行ADF客户端软件:如果正常,则会出现ADF的图形界面。
如果你的这台计算机没有被收录在license许可里,软件会出现如下图,请其中最后二行的内容并发送至662@,软件管理员将回复用户一个新license文件。
二、在ADF客户端软件进行前处理工作1.建立模型设置参数等,保存,会产生一个${JOBNAME}.run文件。
如示例AgI_asoexcit.run2.编辑作业脚本:使用纯文本编辑软件(如记事本)编辑脚本文件,如示例adf_test.pbs。
可以做一个范本,以后要计算不同的任务的时候,只需拷贝到对应目录,稍作修改即可。
具体内容如下:#PBS -j oe#PBS -N M3_2x12_2012#PBS -q adf#PBS -l nodes=2:ppn=12###Apply for the use of two compute nodes, each node of 12 nuclear#PBS -Vcd $PBS_O_WORKDIR### ---------------------------------------### PLEASE MODIFY YOUR JOBNAME### ---------------------------------------JOBNAME=AgI_asoexcit###The name of the job, corresponding to the ‘*’ of *.run### ---------------------------------------### BEGINNING OF EXECUTION### ---------------------------------------export ADFHOME=/public/software/adf2012.01###The root directory of ADFexport SCMLICENSE=$ADFHOME/license.txt###The path of ADF license fileexport SCM_TMPDIR=/public/home/test/adf_tmpe### Make sure you have right to write into this directory. ADF will generate scratch in this ###directory, for this reason, you’d better cleanthis directory termly.export ADFBIN=$ADFHOME/bin###Directory for ADF executable filesexport ADFRESOURCES=$ADFHOME/atomicdata###Basis file and force field file directoryexport PATH=$ADFBIN:$PATH###Add adf executable file derectory into PA THNP=`cat $PBS_NODEFILE|wc -l`###PBS finds appropriate nodes from nodes list to run this job/public/home/test/test/${JOBNAME}.run > ${JOBNAME}.out###${JOBNAME}.run is generated by ADFinput, while ${JOBNAME}.out is output of this job in ###text format. logfile will be generated automatically. After you submit this job, type tail –f ###logfile, you can monitor the task.注意:其中红色字的内容,用户自己根据自己不同的作业修改为相应的内容。

ADF中文教程拉曼光谱的计算拉曼光谱是分析物质结构和化学键的一种非常重要的工具。
与传统的红外光谱相比,拉曼光谱具有高灵敏度、高分辨率和非接触等优点,因此在很多领域,如化学、生物、材料和环境等方面有广泛的应用。
本文将介绍关于拉曼光谱的计算方法。
首先,我们需要了解拉曼效应的原理。
当物质受到激发光的照射时,部分光子会散射并与物质中的分子相互作用,改变了光子的能量。
这种散射光的能量与入射光的能量之差称为拉曼位移。
通过测量散射光的频移,我们可以得到拉曼光谱,从而了解物质的结构和化学键。
在计算拉曼光谱时,最基本的方法是使用密度泛函理论(DFT)计算物质的振动模式。
DFT是一种量子力学的计算方法,通过求解物质中电子的波函数来描述物质的性质。
在拉曼光谱计算中,我们需要计算物质在不同的振动模式下的能量差异。
首先,我们需要准备计算模型。
这包括确定物质的晶胞结构和基元(原子组合)。
对于一些简单的化合物,我们可以从实验数据中获得晶胞结构和基元的信息。
对于复杂的分子,我们需要使用软件工具,如VASP、Quantum Espresso等来构建计算模型。
接下来,我们需要选择合适的DFT方法和基组来进行计算。
常用的DFT方法包括B3LYP、PBE等,而基组可以选择在不同基组下进行计算,以确定计算结果的可靠性。
此外,还需要考虑是否考虑溶剂效应等因素。
一旦计算模型和参数选择好了,我们就可以进行振动模式计算。
一般来说,我们需要计算物质的力常数矩阵,然后通过对角化该矩阵得到振动模式的频率和振动模式。
频率和振动模式揭示了物质中原子的运动方式,可以用来解释拉曼光谱的各个峰值。
最后,我们可以将计算得到的振动频率与实验测定的拉曼光谱进行比较,从而验证计算结果的准确性。
如果计算结果与实验结果不一致,我们可以检查计算模型和参数选择是否正确,如果有问题,可以对计算模型进行调整。
总之,计算拉曼光谱是一项复杂而有挑战性的任务,需要我们掌握一定的量子化学和计算化学知识。

ADF教程如何去掉FCF计算中的虚频和低频模式ADF是一款计算量子化学的软件工具,用于模拟和计算原子和分子系统的性质。
在计算过程中,ADF会生成一个频率矩阵,包含了分子中各个原子的振动频率。
然而,在一些情况下,ADF生成的频率可能包含一些虚频(即负频)和低频振动模式,这些结果可能对计算结果的准确性产生影响。
因此,我们有时需要去掉这些虚频和低频模式,以确保计算结果的准确性。
去除ADF计算结果中的虚频和低频模式通常需要通过一些后处理方法来实现,以下是利用ADF提供的工具和方法进行处理的步骤:1. 首先,我们需要生成原子和分子的振动频率。
在ADF中,可以使用频率分析(Freq)模块进行振动频率的计算。
该模块会生成一个频率矩阵,其中包含了所有原子的振动频率。
2. 生成频率矩阵后,需要查看频率矩阵中的虚频和低频模式。
在ADF的输出文件中通常会包含一个“Vibrational Frequencies”的部分,其中列出了所有振动频率。
需要注意的是,在一些情况下,虚频其实是由于计算误差引起的,因此并非所有的虚频都需要被移除。
3. 接下来,需要对频率矩阵进行处理,去除虚频和低频模式。
在ADF中,我们可以使用VibScale工具来进行频率缩放和模式筛选操作。
VibScale工具可以通过命令行或者ADF输入文件来使用。
4. 首先,我们需要运行VibScale,生成一个输入文件。
输入文件中需要包含要进行处理的频率矩阵文件的路径,并指定计算所需的其他参数,如频率缩放因子等。
可以使用ADF提供的帮助文档来了解更多关于VibScale的使用方法和参数说明。
5. 运行VibScale后,它会生成一个新的频率矩阵文件,其中已经移除了虚频和低频模式。
可以使用ADF中的频率分析工具再次确认新的频率矩阵是否满足要求。
需要注意的是,去除虚频和低频模式可能会对计算结果产生影响。
在一些情况下,虚频和低频模式实际上是系统的稳定振动模式的一部分。

转载+收藏数理化地⽣常⽤软件⼀数学:1、数学软件:(1)常见的通⽤数学软件包包括:Matlab和Mathematica和Maple,其中Matlab以数值计算见长,Mathematica和Maple以符号运算、公式推导见长(2)专⽤数学包包括:绘图软件类:MathCAD,Tecplot,IDL,Surfer,Origin,SmartDraw,DSP2000数值计算类:Matcom,DataFit,S-Spline,Lindo,Lingo,O-Matrix,Scilab,Octave数值计算库:linpack/lapack/BLAS/GERMS/IMSL/CXML有限元计算类:ANSYS, MARC,PARSTRAN, FLUENT, FEMLAB,FlexPDE,Algor,COSMOS, ABAQUS,ADINA数理统计类:GAUSS ,SPSS,SAS, Splus学公式排版类:MathType,MikTeX,ScientificWorkplace,Scientific Nootbook2、数学编程:包括Fortran、C/C++、VB...MatLab、Maple、Mathematica、Femlab、......等编程,讨论各种算法,包括神经⽹络,模拟退⽕等,可以应⽤到计算数学,统计学等。
⼆、物理1、物理软件:1基本⽤途软件(1)符号计算:mathematica:这是唯⼀⼀个商业软件,下⾯有的程序依赖于它,⽽且由于Wolfram当年也是⾼能物理出⾝,因此个⼈觉得该软件的使⽤体验很好,也是我唯⼀动⼼购买正版的软件。
form:⼤规模处理符号表达式的利器,下⾯有的软件包依赖于它,适宜⽤来做⾼圈多腿图的计算,但是⽤起来没有mathematica⽅便。
maxima:这个是mathematica的免费替代品,但缺点是很多表达式没法像mathematica那样化简,不过好在提供源代码.(2)数值计算:gsl:C程序写的数值计算库,内容还⽐较全⾯,⽤来做数值计算很⽅便,⽂档⽐较详细且集中。

化学实验数据处理与分析方法与软件随着科学技术的发展和实验方法的改进,化学实验数据处理与分析变得越来越重要。
为了提高数据处理的准确性和效率,科学家和研究人员开发了许多方法和软件来帮助处理和分析化学实验数据。
本文将介绍一些常用的化学实验数据处理与分析方法与软件。
1. Excel表格处理数据Excel是广泛使用的电子制表软件,也是数据处理和分析的常用工具。
它提供了各种功能和公式,可以帮助用户进行统计、图表、回归等数据处理操作。
在化学实验中,可以使用Excel来整理实验数据、计算平均值和标准偏差、绘制曲线等。
通过合理利用Excel的功能,可以快速准确地处理和分析大量的化学实验数据。
2. Origin数据分析软件Origin是一款专业的数据分析和制图软件,广泛应用于科研领域。
它具有强大的数据处理和分析功能,可以进行曲线拟合、统计分析、参数估计等操作。
Origin还提供了丰富的绘图工具,可以制作各种类型的图表,如散点图、直方图、线图等。
在化学实验中,可以使用Origin来进行数据的曲线拟合和统计分析,从而更好地理解实验结果。
3. Python编程语言与数据处理库Python是一种简单易学的编程语言,广泛应用于数据科学领域。
它提供了许多数据处理和分析的库,如NumPy、Pandas和Matplotlib等。
这些库可以帮助用户高效地进行数据处理、统计分析和数据可视化。
在化学实验中,可以使用Python编程语言和相关库来处理实验数据,进行曲线拟合、统计分析和绘图等操作。
相比其他软件,Python具有灵活性强、可定制性好等优势。
4. 化学实验数据处理方法除了软件工具之外,化学实验数据处理还可以采用各种方法和技巧。
例如,可以计算实验数据的平均值和标准差,以评估实验数据的可靠性。
可以进行相关性分析,判断实验变量之间的关系。
还可以利用数学模型和统计方法来分析实验数据,以获取更深入的信息。
在化学实验数据处理过程中,需要注意数据的准确性、异常值的处理和统计方法选择等。

计算化学的软件工具和数据资源计算化学是一种基于计算机科学的化学研究方法,它利用计算机模拟来预测化学物质的物理化学性质和反应机理。
随着计算机科学和化学研究的迅速发展,计算化学的应用已经得到广泛的推广和应用。
为了更加有效地使用计算化学方法,研究人员需要掌握一些计算化学的软件工具和数据资源,让我们一起来看看这些有用的工具和资源。
1. 分子模拟软件分子模拟软件是计算化学研究的核心工具。
它可以用来模拟分子的运动和反应,预测分子的结构、能量和性质。
常用的分子模拟软件有Gromacs、Amber、LAMMPS、CHARMM等。
这些软件通常需要一定的计算机编程技能,但是它们提供了很强的自由度和控制力,可以满足不同研究需求的要求,同时也是训练计算化学研究人员的基本技能。
2. 密度泛函理论软件密度泛函理论(DFT)是一种计算电子结构的方法,它可以用来预测分子的几何构型、电子能级和电荷分布等。
常用的DFT软件有Gaussian、VASP、Quantum ESPRESSO等。
这些软件通常需要一定的物理、数学和计算机科学知识,但是它们提供了很强的准确性和可靠性,可以用来研究很多重要的化学问题。
3. 虚拟筛选软件虚拟筛选软件是用来寻找化学药物和分子杂交物的软件工具。
它利用基于计算机的分子模拟和化学信息检索技术,可以从大规模化学库中筛选出具有特定生物活性的化合物。
常用的虚拟筛选软件有Autodock、Vina、Glide等。
这些软件允许研究人员进行高通量筛选和分子设计,可以大大提高化学药物和分子杂交物的研发速度和成功率。
除了这些软件工具之外,计算化学研究人员还需要掌握一些数据资源,这些数据资源可以用来支持计算化学研究的可靠性和准确性。
4. 化合物数据库和手册化合物数据库和手册是计算化学研究人员必备的资源之一。
它们收集了大量的化学结构、性质和反应信息,包括分子式、结构式、物理化学性质、毒性信息、化学反应机理等。
常用的化合物数据库和手册有Beilstein、PubChem、ChemSpider、EPA等。

ADF软件特点
ADF模块
1.擅长大分子的计算(目前已经报道大量使用ADF对几千原子的大分子进行DFT计算);
2.擅长过渡金属体系的计算;
3.擅长考虑相对论效应的计算;
4.并行效率非常高;
5.支持GPU加速;
6.软件的生命力强,开发组很活跃;
7.使用Slater基函数,因此对于原子核性质的计算,比Gaussian型基组软件可靠;
8.图形界面友好
BAND模块
9.过渡态搜索非常方便;
10.界面友好;
11.支持固体表面化学反应的化学键分析(即将发布,目前版本也支持部分功能);
12.一维、二维体系计算速度优于流行的平面波程序(因为不需要处理真空层带来的计算量);
13.一维、二维体系的表面溶剂化更合理
14.能够计算自旋轨道耦合;
15.能够进行元素周期表所有元素的全电子计算
ReaxFF模块
1.界面友好;
2.并行效率高于原始ReaxFF;
3.力场更新更多;
4.分析工具丰富(对于化学反应分析,非常必要);
COSMO-RS模块
1.ADF软件包自带分子库以及新增分子库的工具;
DFTB模块
2.大量新的参数;
3.图形界面友好,功能较多(声子、能带、结构优化、过渡态搜索等);
4.分析工具友好易用
5.支持蛋白质大分子的紫外-可见吸收光谱计算
NBO模块
1.计算比原始NBO方便;
2.图形界面比原始NBOView方便。

高三化学学科教学辅助工具推荐化学学科在高中阶段是一门重要的科学学科,对于学生的综合素质提升和未来的学业规划有着重要的影响。
为了帮助高三化学学科学生更好地掌握知识,提高学习效率,教学辅助工具的选择与应用显得尤为重要。
本文将针对高三化学学科教学辅助工具进行推荐,旨在帮助教师和学生选择适合的辅助工具,提升学习效果。
一、电子化学教辅软件随着互联网和科技的发展,电子化学教辅软件以其直观、互动、实践性等特点受到越来越多人的关注和推崇。
以下是几款经典的电子化学教辅软件推荐:1. 化学实验模拟软件化学实验是化学学科的重要组成部分,通过模拟软件可以在实验室环境下进行安全实时的实验观测和操作,提供实验的视觉和实践体验。
2. 化学知识查询软件化学知识查询软件集中了大量的化学知识点,可以根据关键词查询相关的实验原理、性质、反应等内容,方便学生对知识进行快速了解和记忆。
3. 化学反应动画软件通过化学反应动画软件,学生可以观察和了解化学反应的过程,加深对化学反应原理和机理的理解。
二、智能化学教学设备智能化学教学设备是利用先进的科技手段,结合化学学科教学需求,为学生提供实验操作和观察的便利工具。
以下是几种常见的智能化学教学设备推荐:1. 分子模型分子模型是可拆卸并拼接的结构模型,能够直观地展示和模拟化学物质的分子结构,帮助学生理解和记忆化学分子的构成和性质。
2. 智能化学实验箱智能化学实验箱通过智能感应技术和虚拟实验技术,模拟各种化学实验的过程和操作,为学生提供近乎真实的实验环境和实验操作,培养学生的实验思维和实践能力。
三、化学学习平台化学学习平台以其内容丰富、互动性强等特点,成为高三化学学科学习的重要辅助工具。
以下是几个优秀的化学学习平台推荐:1. 在线化学课件平台在线化学课件平台集中了大量的化学知识点和实例,以多媒体的形式呈现,配有相关的图表和动画,增加了学习的趣味性和可理解性。
2. 化学学习网站化学学习网站提供了丰富的学习资源,包括课程视频、学习资料、习题和练习题等,学生可以自主选择合适的学习内容,进行针对性的学习和复习。

EXECL化工计算应用介绍Excel是一款功能强大的电子表格软件,广泛应用于各个领域。
在化工行业中,Excel可以用来进行各种计算和数据处理,帮助化工工程师和研究人员提高工作效率和准确性。
下面将介绍几种常见的Excel化工计算应用。
1.物料平衡计算:物料平衡是化工设计与操作中的基础内容,用于确定化工过程中原料和产物的质量和数量关系。
通过建立一个Excel表格,可以方便地记录和计算物料输入和输出的质量和流量。
同时,可以根据输入数据自动计算物料平衡,并实时更新结果。
例如,可以利用Excel计算一个化工生产过程中的物料收支情况,并进行质量和流量的平衡。
2.热力平衡计算:在化工设计和操作中,热力平衡计算是必不可少的一项任务。
Excel提供了一系列的函数和工具,可以用于热力平衡计算。
可以通过建立一个Excel模型,将输入的能量输入和输出定量化,并通过一系列的公式和计算函数计算热力平衡。
例如,可以利用Excel计算一个化工装置中的热量传递和能量输入,从而优化设备的设计和操作。
3.曲线拟合和数据分析:Excel提供了强大的数据分析功能,可以帮助化工工程师和研究人员对实验数据进行处理和分析。
可以利用Excel的曲线拟合功能,对实验数据进行拟合分析,提取有效的信息和关系。
例如,可以通过拟合实验数据,得到一个反应动力学方程或者设备性能的关系曲线。
此外,Excel还提供了统计分析功能,可以帮助进行数据处理和统计,从而得到更加准确的结果。
4.设备的设计和优化:对于化工工程师来说,设备的设计和优化是一项重要的任务。
Excel可以用于设备的设计计算和参数分析,从而帮助改善设备的性能和效率。
可以通过建立一个Excel模型,输入设备的参数和运行条件,然后利用一系列的计算公式和函数,计算设备的设计要求和参数。
同时,可以通过改变不同参数的数值,进行设备的优化和性能改进。
5.数据可视化和报表生成:Excel提供了很多图表和图形功能,可以将数据可视化,更直观地展示计算结果和数据分析。
常用化学软件大全化学结构式有关化学结构式编辑的软件市面上非常之多,它们各有所长。
既有商品的,亦有对教育界及家用免费的。
其功能主要是描绘化合物的结构式、化学反应方程式、化工流程图、简单的实验装臵图等化学常用的平面图形的绘制。
常见的这类软件有:ChemDraw, ChemWindow, ISIS Draw, ChemSketch等。
前两个为商业软件,有关它们的资料可以查阅各自的网站/ 和/,最新版本分别为6.0和6.5。
后两个对教育界及家用为免费软件,可以在它们各自的网站/和/上下载,最新版本分别为2.2和4.0。
ChemDraw为当前最常用的结构式编辑软件,除了以上所述的一般功能外,其ultra版本还可以预测分子的常见物理化学性质如:熔点、生成热等;对结构按IUPAC原则命名;预测质子及碳13化学位移等。
ChemWindow的一个最突出的特点是与光谱的结合,它的 6.5 Spectroscopy 版本包括了一个约五万张13C NM的数据库(达250兆),因而其预测更加精确;除了根据化合物的结构预测13C NMR化学位移外,还能预测红外图谱、质谱等,更可以读入标准格式的NMR、IR、Raman、UV及色谱图。
这些程序虽然可以画出非常好的二维化学结构,但除了ChemSketch 外,要表现出三维的化学结构则十分困难,必须依赖于一些专门的3D软件来实现。
三维结构比较有名的化学三维结构显示与描绘软件有:Chem3D, WebLab Viewer Pro, RasWin, ChemBuilder 3D, ChemSite等,它们都能够以线图(wire frame), 球棍(ball and stick), CPK及丝带(ribbon)等模式显示化合物的三维结构。
其中的RasWin和WebLab Viewer的Lite版只能显示而无法编辑三维分子模型,为免费软件,RasWin可以在几乎所有的化学软件站点找到,WebLab Viewer的下载地址为/。
计算化学基本概念分子模拟(Molecular Modeling)泛指用于模拟分子或分子体系性质的方法,定位于表述和处理基于三维结构的分子结构和性质。
Quantum Mechanics (QM) 量子力学Molecular Mechanics (MM) 分子力学Theoretical Chemistry 理论化学Computational Chemistry 计算化学Computer Chemistry 计算机化学Molecular Modeling 分子模拟量子化学简介量子化学的研究范围和内容9稳定和不稳定分子的结构、性能,及其结构与性能之间的关系9分子和分子之间的相互作用9分子和分子之间的相互碰撞和相互反应等问题计算与预测各种分子性质(如分子几何构型、偶极矩、分子内旋势能、NMR、振动频率与光谱强度)预测化学反应过程中的过渡态及中间体、研究反应机理理解分子间作用力及溶液、固体中的分子行为计算热力学性质(熵、Gibbs函数、热容等)量子力学与经典力学的差别首先表现在对粒子的状态和力学量的描述及其变化规律上。
在量子力学中,粒子的状态用波函数来描述,它是坐标和时间的复函数。
为了描述粒子状态变化的规律,就需要找出波函数所满足的运动方程。
这个方程是薛定谔在1926年首先提出的,被称为薛定谔方程。
求解薛定谔方程,即可从电子结构层面来阐明分子的能量、性质及分子间相互作用的本质。
Schrödinger 方程The ab initio Molecular Orbital TheoryThe Hartree-Fock EquationThe Self-Consistent Field TheoryLinear Combination of Atomic OrbitalsBasis Sets: Slater-Type Orbitals(STO) and Gaussian-Type Orbitals(GTO) 当我们决定由原子轨道线性组合成分子轨道时,就要考虑采取什么数学形式来表示原子轨道。
计算机化学及化学软件网站大全http://www.chem.ac.ru/Chemistry/Soft/ 化学软件网址列表。
http://antas.agraria.uniss.it/software.html 化学软件列表和FTP站点列表。
http://bogense.chem.ou.dk/~icc/software.html 计算化学软件相关的网址。
http://www.lctn.uhp-nancy.fr/Chercheurs/Xavier.Assfeld/links.html 量化软件,基组库,量化教学。
http://www.sc.ehu.es/powgep99/dcytp/teoricos/txoni/programak.htm 量子化学软件和其它有用的应用程序。
/reports/1996/6802brown/ 量子化学缩写辞典。
.tw/pub/ 台湾高速电脑中心各学科领域研究软件,可以下载。
包括Chemistry,databases,parallel,physics,visualization。
/~pac/reps.html Clarkson大学相对论有效势数据库。
提供第二至第六周期全部元素的相对论有效势基组下载,可以用在Gaussian和COLUMBUS上。
/quantum/ 有Dyall博士的《相对论量子化学》授课笔记下载,即将出书。
/ Computational Chemistry List,有免费量化软件下载,以及量子化学论坛等。
/Doc/publist.html 与ADF软件有关的一些博士论文。
从中可以学到很多量化知识。
http://leroy.uwaterloo.ca/ 处理双原子分子光谱参数的一些fortran源程序(LEVEL,BCONT,RKR1,DSParFit)。
申请后,就会发到你的E-mail信箱。
http://zeus.polsl.gliwice.pl/~nikodem//linux4chemistry.html 集中了绝大多数用在Linux下的化学软件。
ADF软件包介绍ADF:非周期体系的密度泛函计算模块ADF是历史最悠久的模块,擅长重元素体系、有机体系的精确计算,包括电荷转移、荧光、磷光、化学键分解、片段分析、轨道组分分析等功能。
高度并行化(上千核并行),多种解决收敛问题的机制,丰富的性质计算与图形显示功能是ADF的独特优势。
让电子结构计算、催化与化学性质的理解变得简单直观。
另外,ADF的泛函库非常庞大,最新发表的重要泛函会很快地被纳入ADF的泛函库。
(详细介绍)BAND:周期性体系的密度泛函计算模块与VASP等采用平面波基组的程序有所不同,BAND采用数值基组与Slater基组结合,能够处理真正意义上的二维和一维体系,因此而对表面进行溶剂化效应的模拟。
同时不必担心赝势的精确度对不同体系的可靠性问题。
BAND能够对元素周期表中所有元素进行精确第一性原理计算。
在表面催化方面,精确度和易用性非常高。
(详细介绍)DFTB:近似密度泛函计算模块DFTB是在DFT计算的库仑积分过程中进行了参数化,因此大大地提高了密度泛函的计算效率。
在DFTB官方发布的参数之外,ADF基于QUASINANO项目开发了更多参数。
考虑长程相互作用的色散校正与新DFTB3方法可以精确地处理带电体系。
使用普通台式机或笔记本就可以实现大体系、大时间尺度的单点计算、结构优化、过渡态计算、频率计算,以及分子动力学模拟。
(详细介绍)ReaxFF:反应力场ReaxFF通过力场的方式,模拟介观尺度下的化学反应,以及有关的热力学、动力学性质。
在过去几十年里用于各种不同反应体系模拟,包括:溶液环境、界面、金属及金属氧化物表面分子反应,并支持Berendsen温度velocity Verlet的NVT、NPT和NVE系综的动力学计算功能。
在有机体系(如有机分子的燃烧、高温热解等)、金属催化、合金材料氧化、炸药爆炸等各种工程问题的模拟方面取得了很大的成功。
(详细介绍)MOPAC2012:半经验电子结构计算经验参数化的量子化学方法。