总氰化物测定方法
- 格式:doc
- 大小:49.00 KB
- 文档页数:13

水质氰化物的测定分光光度法作业指导书警告:氰化物属于剧毒物质,操作时应氨规定佩戴防护器具,避免接触皮肤和衣服;检测后的残渣残液应做妥善的安全处理。
1参考标准《水质氰化物的测定容量法和分光光度法》HJ484-2009方法2 异烟酸-吡唑啉酮分光光度法2适用范围本规程适用于地表水、生活污水和工业废水采用异烟酸-吡唑啉酮比色法测定水质中的总氰化物。
最低检出浓度为0.004mg/L;测定下限为0.016mg/L,测定上限为0.25mg/L。
3定义3.1总氰化物是指在磷酸和EDTA存在下,于pH<2介质中,加热蒸馏,能形成氰化氢的氰化物,包括全部简单氰化物(多为碱金属和碱土金属的氰化物、铵的氰化物)和绝大部分络合氰化物,但不包括钴氰络合物。
3.2 易释放氰化物在pH=4介质中,硝酸锌存在下,加热蒸馏,形成氰化氢的氰化物,包括全部简单氰化物(多为碱金属和碱土金属的氰化物、铵的氰化物)和锌氰络合物,不包括铁氰化物、亚铁氰化物、铜氰络合物、镍氢络合物、钴氰络合物。
4原理4.1蒸馏原理4.1.1总氰化物:向水样中加入磷酸和EDTA二钠,在pH<2条件下,加热蒸馏,利用金属离子和EDTA络合能力比和氰离子络合能力强的特点,使络合氰化物离解出氰离子,并以氰化氢形式被蒸馏出,用氢氧化钠溶液吸收。
4.1.2 易释放氰化物:向水样中加入酒石酸和硝酸锌,在pH=4条件下,加热蒸馏,简单氰化物和部分络合氰化物以氰化氢形式被蒸馏出,用氢氧化钠溶液吸收。
4.2 反应原理在中性条件下,样品中的氰化物和氯胺T反应生成氯化氰,再和异烟酸作用,经水解后生成戊烯二醛,最后和吡唑啉酮缩合生成蓝色染料,其颜色和氰化物含量成正比,在638nm波长进行光度测定。
4仪器4.1分光光度计;4.225ml具塞比色管;4.3500ml全玻璃蒸馏器;4.4100ml量筒或容量瓶;4.5600W或800W可调电炉;5药品及试剂测定过程中,只使用公认的分析纯试剂和不含氰化物和活性氯的蒸馏水或具有同等纯度的水。

氰化物实验作业指导书(共14页) -本页仅作为预览文档封面,使用时请删除本页-总氰化物的测定1、方法依据水质总氰化物的测定异烟酸-吡唑啉酮分光光度法HJ484-20092、适用范围本方法适用于地表水、生活污水和工业废水中氰化物的测定。
本方法检出限为L,测定下限为L,测定上限L。
3、测定原理总氰化物的测定在中性条件下,样品中的氰化物与氯胺T反应生成氯化氰,再与异烟酸作用,经水解后生成戊烯二醛,最后与吡唑啉酮缩合生成蓝色染料,在波长638nm出测量吸光度。
4、干扰和消除活性氯等氧化物干扰测定试样中存在与活性氯等氧化物干扰测定,可在蒸馏前加亚硫酸钠溶液(Na2SO3)排除干扰。
亚硝酸离子干扰测定试样中存在亚硫酸离子干扰测定,可在蒸馏前加亚硫酸钠溶液(Na2SO3)排除干扰。
硫化物干扰测定试样中存在硫化物干扰测定,可在蒸馏前加碳酸镉(CdCO3)或碳酸铅(PbCO3)固体粉末排除干扰。
油类物质干扰测定少量油类对测定无影响,中性油或酸性油大于40mg/L时干扰测定,可加入水样体积的20%量的正己烷(C6H14),在中性条件下段时间萃取,分理处正己烷相后,水相用预蒸馏测定。
5、试剂本标准所用试剂除非另有说明,分析时均用使用符合国家标准的分析纯化学试剂,实验用水为新制备的不含氰化物和活性氯的蒸馏水或去离子水。
氰化钾标准溶液氰化钾贮备溶液的配置和标定:称取氰化钾(KCN,注意剧毒!避免尘土的吸入或与固体或溶液的接触)于100mL棕色容量瓶中,溶于氢氧化钠并稀释至标线,摇匀,避光贮存于棕色瓶中,4℃以下冷藏至少可稳定2个月。
本溶液氰离子(CN-)质量浓度约为1g/L,临用前用硝酸银标准溶液标定其准确浓度。
氰化钾贮备溶液的标定:吸取氰化钾贮备溶液于锥形瓶中,加入50mL水和1mL氢氧化钠,加入试银灵指示剂,用硝酸标准溶液滴定至溶液有黄色刚便为橙红色为止,记录硝酸银标准溶液用量(V1)。
另取实验用水做空白实验,记录硝酸银标准溶液用量(V0)。

检测氰化物的方法
1. 铁盐法:将待测物加入铁盐的溶液中,如果有氰化物存在,则会发生氰化反应,生成蓝色的亚铁氰化物。
2. 火焰试验法:将待测物添加到火焰中,如果有氰化物存在,则会出现蓝绿色的火焰。
3. 银镜反应法:将待测物与银盐反应,如果有氰化物存在,则会生成镜面银。
4. 转移分析法:将待测物分别加入稀硝酸和氢氧化钠的溶液中,然后将两种溶液混合,如有氰化物存在,则会出现可见的白色沉淀。
5. 毒蛋白法:将待测物加入一种含有毒性的蛋白质溶液中,如果有氰化物存在,则会导致蛋白质失去活性而不能杀死细菌。
注意:在进行氰化物检测时,应采取正确的保护措施,避免接触到有毒的氰化物。

总氰化物浓度的测定标准曲线的绘制(1)用分析天平准确称取0.2503g分析纯氰化钾溶于100mL水中,则此溶液1mL 相当1mg的CN—标准贮备液。
(空烧杯:45.5675g,总重:45.8178g)(2)取标准贮备液2.5mL,用250mL容量瓶定容到100mL,此为标准中间液。
(3)取标准中间液2.5mL,用25mL比色管稀释成25mL溶液,此为标准使用液。
(4)分别取标准适用液0、0.3、1、2、3、4、5mL于25mL比色管中。
(5)加少量蒸馏水,加入1~2滴醋酸酸化,加饱和溴水1~2滴呈现黄色不退,摇匀静置10分钟。
(6)加数滴0.5%硫酸肼至黄色褪去再加过量一滴,摇匀,加3mL吡啶联苯胺溶液,定容至10mL,摇匀,静置15分钟。
(7)于520波长下测定吸光度,根据数据绘制标准曲线。
总氰化物浓度的测定原理:溶液中的CN与饱和溴水反应生成溴化氰,再与吡啶联苯胺反应生成不同色度的紫红色染料,在520纳米处有最大吸光度。
本方法最低检出浓度为0.05毫克每升。
测定上限为10毫克每升。
主要试剂及仪器:冰醋酸:3:7溴水:先加入小量溴素,再加入水即可硫酸肼溶液:0.5%吡啶联苯胺溶液(显色剂)(60ml配置方法):取0.5克联苯胺容于10ml浓度为2%盐酸中并加热,后取50ML浓度为60%的吡啶溶液氰根标准溶液:取0.2503g分析纯氰化钾溶于100mL水中,则此溶液1mL相当1mg 的CN-标准溶液。
25mL具塞比色管、721比色分光光度计步骤:(1)取过滤后水样1~5mL于25mL比色管中,加少量蒸馏水,加入1~2滴醋酸酸化,加饱和溴水1~2滴呈现黄色不退,摇匀静置10分钟。
(2)加数滴0.5%硫酸肼至黄色褪去再加过量一滴,摇匀,加3mL吡啶联苯胺溶液,定容至10mL,摇匀,静置15分钟。
(3)于520波长下测定吸光度。
(4)把数值带入总氰标准曲线回归方程计算出水样浓度。
图 4 总氰标准曲线注:若浓度过高,则将水样稀释100或500倍,然后计算出浓度。


挥发酚和总氰化物在三种水质中测定方法的比对挥发酚和总氰化物是水质检测中常见的重金属污染物。
测定这两种污染物的方法对于监测水质的安全性和环境保护具有重要的意义。
本文将对挥发酚和总氰化物在三种水质中测定方法进行比对,分析其优缺点并提出改进建议。
一、挥发酚的测定方法比对1. 分光光度法分光光度法是一种常见的挥发酚测定方法,其原理是利用挥发酚与Fe3+形成有色络合物,然后根据络合物的吸光度来测定挥发酚的含量。
这种方法简单、快速、灵敏度高,适用于水样中挥发酚的测定。
2. 气相色谱法气相色谱法是一种高灵敏度、高分辨率的测定方法,可以对挥发酚进行准确的定量分析。
其原理是通过气相色谱仪对样品中的挥发酚进行分离和检测,具有高准确性和精确度。
3. 液-液萃取法液-液萃取法是一种传统的测定方法,其原理是通过有机溶剂将水样中的挥发酚萃取出来,然后再进行分析。
这种方法操作简单,适用于对挥发酚进行初步筛查和快速测定。
1. 降解-熏蒸法降解-熏蒸法是一种常见的总氰化物测定方法,其原理是先将水样中的氰化物氧化成氰化气,然后使用熏蒸法将氰化气转化为可测的化合物进行定量分析。
这种方法具有高灵敏度和准确性,适用于对总氰化物进行快速测定。
2. 氨氮法三、三种测定方法比对分析从以上测定方法的比对可以看出,不同的测定方法具有各自的优缺点。
分光光度法和气相色谱法具有高灵敏度、高准确性和精确度,适用于对挥发酚和总氰化物进行准确的定量分析。
这两种方法的操作较为繁琐,需要较长的分析时间。
液-液萃取法和氨氮法操作简单,适用于对挥发酚和总氰化物进行初步筛查和快速测定,但是其灵敏度和准确性较低。
降解-熏蒸法和高效液相色谱法则是综合了灵敏度和操作简便性的测定方法,适用于对挥发酚和总氰化物进行快速、准确的测定。
四、改进建议针对以上测定方法的比对分析,可以提出一些改进建议。
在实际应用中可以根据不同的需求选择合适的测定方法,如对于要求高灵敏度和准确性的测定,可以优先选择分光光度法和气相色谱法;对于快速筛查和初步测定,可以考虑使用液-液萃取法和氨氮法。

BCK盐1.准确称取m(g)样品,融于200ml蒸馏水中,移入500ml的蒸馏瓶中(若氰化物含量高,可少取样品),加数粒玻璃珠。
2.往接收瓶内加入10ml1%氢氧化钠溶液,作为接收液。
3.馏出液导管上端接冷凝管的出口,下端插入接收瓶的吸收液中,检查连接部位,使其严密。
4.将10ml10%EDTA二钠溶液加入蒸馏瓶内。
5.迅速加入10ml磷酸,但样品碱度大时可适当多加磷酸,使pH小于2,立即连接好装置,打开冷凝水,打开加热器由低档逐渐升高加热蒸馏。
6.接收瓶内溶液接近70ml时,停止蒸馏,用少量水冲洗馏出液导管,取出接收瓶,用水稀释至标线,此馏出液待测定总氰化物。
7.取100ml馏出液于锥形瓶中,调节pH>11。
8.加入3~5滴试银灵指示剂,用硝酸银标准溶液滴定至溶液由黄色变为橙红色为止,记下读数v,同时做空白。
9.计算方法:x=c×(v-v0)×52.04×1000/m式中:c----------硝酸银标准溶液浓度,mol/lv----------测定试样时硝酸银标准溶液用量,mlv0---------空白试验硝酸银标准溶液用量,mlm----------样品质量,g复合肥中CN检测方法10.准确称取m(g)样品,用200ml蒸馏水分多次将样品加入500ml的凯氏瓶中(若氰化物含量高,可少取样品),加数粒玻璃珠。
11.往接收瓶内加入10ml1%氢氧化钠溶液,作为接收液。
12.将10ml10%EDTA二钠溶液加入蒸馏瓶内。
13.迅速加入10ml磷酸,但样品碱度大时可适当多加磷酸,使pH小于2,立即连接好装置,打开冷凝水,打开加热器由低档逐渐升高加热蒸馏。
14.接收瓶内溶液接近70ml时,停止蒸馏,用少量水冲洗馏出液导管,取出接收瓶,此馏出液待测定总氰化物。
15.馏出液倒入锥形瓶中,调节pH>11。
16.加入3~5滴试银灵指示剂,用硝酸银标准溶液滴定至溶液由黄色变为橙红色为止,记下读数v,同时做空白。
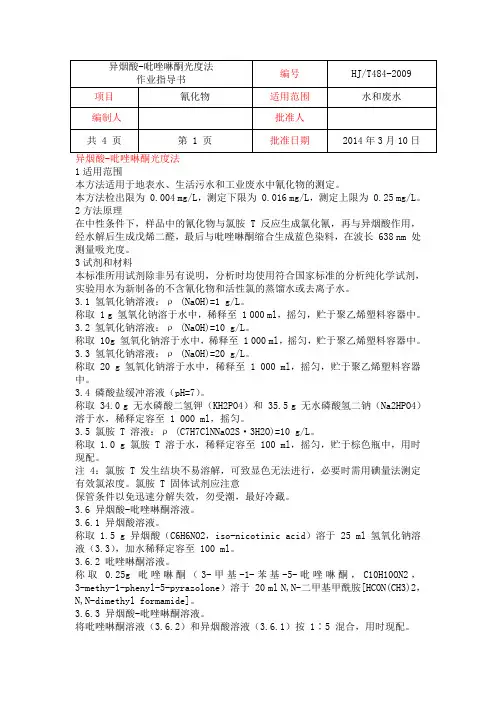
1适用范围本方法适用于地表水、生活污水和工业废水中氰化物的测定。
本方法检出限为 0.004 mg/L,测定下限为 0.016 mg/L,测定上限为 0.25 mg/L。
2方法原理在中性条件下,样品中的氰化物与氯胺 T 反应生成氯化氰,再与异烟酸作用,经水解后生成戊烯二醛,最后与吡唑啉酮缩合生成蓝色染料,在波长 638 nm 处测量吸光度。
3试剂和材料本标准所用试剂除非另有说明,分析时均使用符合国家标准的分析纯化学试剂,实验用水为新制备的不含氰化物和活性氯的蒸馏水或去离子水。
3.1 氢氧化钠溶液:ρ (NaOH)=1 g/L。
称取 1 g 氢氧化钠溶于水中,稀释至 1 000 ml,摇匀,贮于聚乙烯塑料容器中。
3.2 氢氧化钠溶液:ρ (NaOH)=10 g/L。
称取 10g 氢氧化钠溶于水中,稀释至 1 000 ml,摇匀,贮于聚乙烯塑料容器中。
3.3 氢氧化钠溶液:ρ (NaOH)=20 g/L。
称取 20 g 氢氧化钠溶于水中,稀释至 1 000 ml,摇匀,贮于聚乙烯塑料容器中。
3.4 磷酸盐缓冲溶液(pH=7)。
称取 34.0 g 无水磷酸二氢钾(KH2PO4)和 35.5 g 无水磷酸氢二钠(Na2HPO4)溶于水,稀释定容至 1 000 ml,摇匀。
3.5 氯胺 T 溶液:ρ (C7H7ClNNaO2S·3H2O)=10 g/L。
称取 1.0 g 氯胺 T 溶于水,稀释定容至 100 ml,摇匀,贮于棕色瓶中,用时现配。
注 4:氯胺 T 发生结块不易溶解,可致显色无法进行,必要时需用碘量法测定有效氯浓度。
氯胺 T 固体试剂应注意保管条件以免迅速分解失效,勿受潮,最好冷藏。
3.6 异烟酸-吡唑啉酮溶液。
3.6.1 异烟酸溶液。
称取 1.5 g 异烟酸(C6H6NO2,iso-nicotinic acid)溶于 25 ml 氢氧化钠溶液(3.3),加水稀释定容至 100 ml。

水质氰化物的测定-1(蒸馏前处理)氰化物分类:①总氰化物:简单氰化物+绝大多数络合氰化物(铁氰络合物等);②易释放氰化物:简单氰化物+少部分络合氰化物(如锌氰络合物)。
蒸馏前处理的意义:去除干扰离子影响;解离氰络合物,富集提纯氰化物。
一、蒸馏原理1.总氰化物:向水样中加入磷酸和EDTA二钠,在pH<2条件下,加热蒸馏,利用金属离子与EDTA络合能力比与氰离子络合能力强的特点,使络合氰化物离解出氰离子,并以氰化氢形式被蒸馏出,用氢氧化钠溶液吸收。
(不含钴氰络合物,难以解离)2.易释放氰化物:向水样中加入酒石酸和硝酸锌,在pH=4条件下,加热蒸馏,简单氰化物和部分络合氰化物(如锌氰络合物)以氰化氢形式被蒸馏出,用氢氧化钠溶液吸收。
二、实验流程三、注意事项1、水样水样采集和贮存①采集:水样放于聚乙烯塑料瓶或硬质玻璃瓶中,加氢氧化钠至pH>12固定。
②贮存:采样后及时分析。
若不能及时分析,样品应于4℃冷藏,24h内测定。
③固定前硫离子处理:若水样中有大量硫化物,先加碳酸铅粉末除去硫化物,再加氢氧化钠固定(碱性条件下,氰离子与硫离子会形成硫氰酸离子)。
可用乙酸铅试纸检测。
2、水样预蒸馏前离子干扰处理①活性氯等氧化剂:在蒸馏时,氰化物会被分解,使结果偏低。
去除方法:碘化钾-淀粉试纸检验,亚硫酸钠溶液除去。
②亚硝酸离子:亚硝酸盐与EDTA在蒸馏加热条件下,会生成氰化物。
去除方法:加入氨基磺酸溶液,分解亚硝酸盐。
③油类:中性或酸性油类(>40mg/L),可能使蒸馏液变浑浊。
去除方法:可用水样体积20%量的正己烷,中性萃取,水相用于蒸馏。
④醛类:蒸馏加热条件下,醛类和氰化物形成氰醇类(甲醛>0.5mg/L)。
去除方法:加硝酸盐和EDTA。
3、蒸馏①总氰化物:使用EDTA是为了络合金属离子,促进氰络合物分解离。
②易释放氰化物:使用酒石酸(弱酸)调节pH,加入硝酸锌可以抑制铁氰络合物等的解离,但无法抑制锌氰络合物的解离。

氰化物的测定方法
氰化物的测定方法通常包括以下几种:
1. 重铁法:采用硫氰化钾与重铁溶液反应生成硫氰化铁,然后用二硫代硫酸钠溶液测定生成的硫氰根离子的浓度。
2. 电化学法:利用氰化物在电极表面的还原或氧化反应,通过测定电流或电位的变化来确定氰化物的浓度。
3. 光度法:利用氰化物与铁离子生成氧合物复合物,然后利用紫外-可见光谱仪测定复合物的吸光度变化。
4. 气相色谱法:利用气相色谱仪分离氰化物,并通过检测器测定其浓度。
这些方法可以根据具体的实验条件和要求选择合适的测定方法。

全自动流动注射分析仪总氰化物分析方法(异烟酸-巴比妥酸方法)(2-200)µg/L1.1 方法原理本方法原理为:首先样品溶液在pH <2的酸性环境中,在145℃温度条件下和紫外灯开启的前提下,通过在线消解可将溶液中络合的氰化物消解为简单游离氰化物;然后通过在线蒸馏途径可将简单氰化物以气态氰化氢形式蒸馏出来,从而使其与其它干扰物分离开;接下来气态的氢氰酸通过气体渗透膜后,氢氧化钠溶液将其吸收并再进样,在中性条件下,氰离子与氯胺T的活性氯反应生成氯化氰,氯化氰与异烟酸发生反应生成戊烯二醛,戊烯二醛与两个巴比妥酸分子缩合生成红紫色染料,最后的反应溶液通过流通池在600nm处进行比色检测。
1.2 方法模块主要参数及实验室需求操作环境温度:15 ℃-30 ℃相对湿度:≤75%模块电源:交流220V±22V,50Hz±1Hz。
模块功率:约300W噪声:小于70 db防火:拥有防火设备通风:有良好的通风环境位置:远离高电磁干扰远离高振动设备1.3 方法主要指标线性范围:2μg/L CN--200μg/L CN-检出限(DL):0.26μg/L CN-重复性(RSD):≦2%样品分析频率:16样/小时1.4分析单元流程图图 1 总氰化物分析单元流程图1.5 方法主要测试参数主要测试参数可以在进样程序中直接进行设置,进样程序详见下表:表1总氰化物分析方法进样程序设置对象参数设置对象参数洗针时间10注射时间80进样时间140出峰时间30进载流时间30峰宽50到达阀时间200样品周期时间220蠕动泵转速35设定温度1 165设定温度2 60紫外灯卤素灯1.样品保存及前处理2.1 样品采集保存方法水样采集时,首先检验是否存在硫化物干扰(见2.2.1.),如果没有硫化物干扰,应立即加氢氧化钠固定,一般每升水加0.5g固体氢氧化钠,当水样酸度较高时,应多加固体氢氧化钠,保证样品的pH>12,初步处理好样品后,将样品存于聚乙烯塑料瓶或硬质玻璃瓶中,置于暗处存放,避免紫外光的照射。
水质总氰化物测定操作规程一、实验目的:1.了解水体中氰化物的含量,评估水质的污染程度;2.掌握水质总氰化物测定的操作方法,以便进行水质监测和评估。
二、实验仪器和试剂:1.仪器:紫外可见分光光度计、电子天平、离心机等;2.试剂:氰化钠标准溶液、硝酸钠标准溶液、纳氢氨、硫酸酚酞等。
三、实验步骤:1.准备工作:1.1仪器校准:确保分析仪器处于工作状态,对分光光度计进行零点校准;1.2标准曲线制备:采用不同浓度的氰化钠标准溶液,制备出一系列含量递增的标准溶液。
2.水样处理:2.1采样:选择代表性的水样,避免污染和异物干扰,并记录采样点、时间以及其他相关信息;2.2采样容器处理:用硝酸钠标准溶液冲洗采样容器,避免污染和样品损失;2.3过滤:将水样过滤,去除悬浮物;2.4pH调整:根据水样的pH,适当进行调整,使其处于较为稳定的酸性或碱性条件下。
3.样品测定:3.1试剂配置:按照实际需求,配置需要的试剂,如纳氢氨溶液、硫酸酚溶液等;3.2操作流程:3.2.1取一定体积的水样,加入适量的纳氢氨溶液进行缓冲;3.2.2加入一定体积的硫酸酚溶液,用硫酸酚与氰化物发生反应,生成红色化合物;3.2.3实时监测吸光度:将待测溶液放入分光光度计中,选择合适的波长,记录吸光度;3.2.4制备标准工作曲线:将一系列不同浓度的氰化钠标准溶液进行同样操作,记录吸光度;3.2.5计算氰化物含量:根据标准曲线,将实时监测吸光度值与标准曲线进行对照,计算样品中氰化物的含量。
四、实验数据处理:1.标准曲线绘制:根据一系列不同浓度的氰化钠标准溶液的吸光度值,绘制标准曲线;2.水样测定:根据实验操作及实时监测结果,计算水样中氰化物的含量;3.结果分析:根据实验结果,评估水质的污染程度,并进行数据比较、统计和报告撰写。
五、实验安全注意事项:1.操作时戴上防护手套、口罩、护目镜等;2.注意试剂的储存和使用过程中的安全性;3.禁止直接接触有毒、易燃物质;4.实验结束后要彻底清洗仪器和容器。
1FHZHJSZ0024 水质总氰化物的测定F-HZ-HJ-SZ-0024 水质氰化物的测定第一部分总氰化物的测定氰化物属于剧毒物在操作氰化物及其溶液时要特别小心避免沾污皮肤和眼睛吸取溶液一定要用安全移液管或用洗耳球吸溶液切勿吸入口中除氰化物剧毒外吡啶也具有毒性应注意安全使用氰化物可能以氰氢酸氰离子和络合氰化物的形式存在于水中这些氰化物可作为总氰化物和氰化物分别加以测定本方法适用于饮用水地面水生活污水和工业废水活性氯等氧化物干扰使结果偏低可在蒸馏前加亚硫酸钠溶液排除干扰见6.1.7a 硫化物干扰可在蒸馏前加碳酸铅或碳酸镉排除干扰见6.1.7c 亚硝酸离子干扰可在蒸馏前加适量氨基磺酸排除干扰见6.1.7b 少量油类对测定无影响中性油或酸性油大于40mg/L时干扰测定可加入水样体积的20量的正已烷在中性条件下短时间萃取排除干扰本方法分四篇第一篇氰化氢的释放和吸收第二篇硝酸银滴定法第三篇异烟酸吡唑啉酮比色法第四篇吡啶巴比妥酸比色法硝酸银滴定法最低检测浓度为0.25mg/L检测上限为100mg/L 异烟酸吡唑啉酮比色法最低检测浓度为0.004mg/L检测上限为0.25mg/L 吡啶巴比妥酸比色法最低检测浓度为0.002mg/L用72型分光光度计吸光度为0.020左右检测上限为0.45mg/L10mm比色皿0.15mg/L30mm比色皿第一篇氰化氢的释放和吸收定义总氰化物是指在磷酸和EDTA存在下pH2介质中加热蒸馏能形成氰化氢的氰化物包括全部简单氰化物多为碱金届和碱土金属的氰化物铵的氰化物和绝大部分络合氰化物锌氰络合物铁氰络合物镍氰络合物铜氰络合物等不包括钴氰络合物原理向水样中加入磷酸和EDTA二钠在pH2条件下加热蒸馏利用金属离子与EDTA络合能力比与氰离子络合能力强的特点使络合氰化物离解出氰离子并以氰化氢形式被蒸馏出用氢氧化钠吸收试剂测定过程中只能使用公认的分折纯试剂和不含氰化物和活性氯的蒸馏水或具有同等纯度的水3.1 磷酸H3PO41.69g/mL 3.2 10g/L氢氧化钠NaOH溶液 3.3 100g/L EDTA二钠溶液3.4 乙酸铅试纸称取5g乙酸铅PbC2H3O223H2O溶于水中并稀释至100mL将滤纸条浸入上述溶液中1h后取出晾干盛于广口瓶中密塞保存23.5 碘化钾淀粉试纸称取1.5g可溶性淀粉用少量水搅成糊状加入200mL沸水混匀放冷加0.5g碘化钾和0.5g碳酸钠用水稀释至250mL将滤纸条浸渍后〕隽栏墒⒂谧厣 恐忻苋 4?3.6 l5硫酸溶液3.7 亚硫酸钠Na2SO3溶液12.6g/L 3.8 氨基磺酸NH2SO3Hsulfamic acid 3.9 氢氧化钠NaOH溶液40g/L 仪器 4.1 500mL全玻璃蒸馏器4.2 600W或800W可调电炉 4.3 100mL量筒或容量瓶4.4 仪器装置如图所示总氰化物蒸馏装置团可调电炉蒸馏瓶冷凝水出口水接收瓶馏出液导管采样和样品 5.1 采集水样时必须立即加氢氧化钠固定一般每升水样加0.5g固体氢氧化钠当水样酸度高时应多加固体氢氧化钠使样品的pH12并将样品存于聚乙烯塑料瓶或硬质玻璃瓶中5.2 当水样中含有大量硫化物时应先加碳酸镉CdCO3或碳酸铅PbCO3固体粉末除去硫化物后再加氢氧化钠固定否则在碱性条件下氰离子和硫离子作用形成硫氰酸离子而干扰测定注检验硫化物方法可取1滴水样或样品放在乙酸铅试纸3.4上若变黑色硫化铅说明有硫化物存在5.3 如果不能及时测定样品采样后应在24h内分析样品必须将样品存放在冷暗的冰箱内3操作步骤 6.1 氰化氢的释放和吸收6.1.1 量取200mL样品移入500mL蒸馏瓶2中若氰化物含量高可少取样品加水稀释至200mL加数粒玻璃珠6.1.2 往接收瓶4内加入10mL氢氧化钠溶液3.2作为吸收液当样品中存在亚硫酸钠和碳酸钠时可用4氢氧化钠溶液3.9作为吸收液6.1.3 馏出液导管5上端接冷凝管的出口下端插入接收瓶4的吸收液中检查连接部位使其严密6.1.4 将10mLEDTA二钠溶液3.3加入蒸馏瓶2内6.1.5 迅速加入10mL磷酸3.1当样品碱度大时可适当多加磷酸使pH小于2立即盖好瓶塞打开冷凝水打开可调电炉由低档逐渐升高馏出液以24mL/min速度进行加热蒸馏6.1.6 接收瓶4内溶液近100mL时停止蒸馏用少量水洗馏出液导管5取出接收瓶4用水稀释至标线此碱性馏出液A待测定总氰化物用6.1.7 干扰物的排除a. 若样品中存在活性氯等氧化剂由于蒸馏时氰化物会被分解使结果偏低干扰测定可量取两份体积相同的样品向其中一份样品投加碘化钾淀粉试纸3.5l3片加硫酸3.6酸化用亚硫酸钠溶液3.7滴至碘化钾淀粉试纸由蓝色变为无色为止记下用量另一份样品不加试纸仅加上述用量的亚硫酸钠溶液然后按步骤6.1.1至6.1.6操作 b. 若样品中含有大量亚硝酸离子将干扰测定可加入适量的氨基磺酸3.8分解亚硝酸离子一般1mg亚硝酸离子需要加2.5mg氨基磺酸3.8然后按步骤6.1.1至6.1.6操作 c. 若样品中有大量硫化物存在将200mL样品5.2过滤沉淀物用1氢氧化钠3.2洗涤合并滤液和洗涤液然后按步骤6.1.1至6.1.6操作6.2 空白试验用实验用水代替样品按步骤6.1.1至6.1.5操作得到空白试验馏出液B待测定总氰化物用第二篇硝酸银滴定法原理经蒸馏得到的碱性馏出液A用硝酸银标准溶液8.4滴定氰离子与硝酸银作用生成可溶性的银氰络合离子AgCN2-过量的银离子与试银灵指示剂8.1反应溶液由黄色变为橙红色试剂8.1 试银灵指示剂称取0.02g试银灵对二甲氨基亚卞基罗丹宁poradimethylaminobenza1rhodanine溶于100mL丙酮中贮存于棕色瓶并于暗处可稳定一个月8.2 铬酸钾K2CrO4指示剂称取10g铬酸钾溶于少量水中滴加硝酸银标准溶液8.4至产生橙红色沉淀为止放置过夜后过滤用水稀释至100mL 8.3 氯化钠标准溶液0.01mol/L 将氯化钠置瓷坩埚内经500600灼烧至无爆烈声后在干燥器内冷却称取0.5844g于烧杯中用水溶解移入1000mL容量瓶稀释至标线混合摇匀8.4 硝酸银标准溶液0.01mol/L 8.4.1 称取1.699g硝酸银溶于水中稀释至1000mL贮于棕色试剂瓶中格匀待标定后使4用8.4.2 硝酸银溶液的标定a. 吸取0.01mol从氯化钠标准溶液8.310.00mL于150mL具柄瓷皿或锥形瓶9.2中加50mL水同时另取一具柄瓷皿或锥形瓶9.2加入60mL水作空白试验b. 向溶液中加入35滴铬酸钾指示剂8.2在不断搅拌下从滴定管加入待标定的硝酸银溶液8.4.1直至溶液由黄色变成浅砖红色为止记下读数V同样滴定空白溶液读数V0 硝酸银浓度c1mol/L按式1计算100.1001…………………………………………??×VVcc 式中c 氯化钠标准溶液浓度mol/L V 滴定氯化钠标准溶液时硝酸银溶液用量mL V0滴定空白溶液时硝酸银溶液用量mL 8.5 硝酸银标准溶液0.001mol/L 仪器9.1 10mL棕色酸式滴定管9.2 150mL具柄瓷皿或250mL锥形瓶操作步骤10.1 测定10.1.1 取100mL馏出液A如试样中氰化物含量高时可少取试样用水稀释至100mL于具柄瓷皿或锥形瓶9.2中10.1.2 加入0.2mL试银灵指示剂8.1摇匀用硝酸银标准溶液8.4滴定至溶液由黄色变为橙红色为止记下读数Va 10.2 空白试验另取100mL空白试验馏出液B于锥形瓶9.2中按10.1.2进行滴定记下读数V0 注若样品氰化物浓度小于1mg/L可用0.001mol/L硝酸银标准溶液8.5滴定11 结果计算总氰化物含量c2mg/L以氰离子CN-计按式2计算2100004.522102…………………………………………××??VVVVVcca 式中c硝酸银标准溶液浓度mol/L Va测定试样时硝酸银标准溶液用量mL V0空白试验硝酸银标准溶液用量mL V1试样馏出液A的体积mL V2试份测定试样时所取馏出液A的体积mL 52.04相当于1L的1mol/L硝酸银标准溶液的氰离子2CN-质量g 第三篇异烟酸吡唑啉酮比色法原理在中性条件下样品中的氰化物与氯胺T反应生成氯化氰再与异烟酸作用经水解后生成戊烯二醛最后与吡唑啉酮缩合生成蓝色染料其颜色与氰化物的含量成正比5试剂13.1 20g/L氢氧化钠溶液13.2 1g/L氢氧化钠溶液13.3 磷酸盐缓冲溶液pH7 称取34.0g无水磷酸二氢钾KH2PO4和35.5g无水磷酸氢二钠Na2HPO4于烧杯内加水溶解后稀释至1000mL摇匀放入试剂瓶存于冰箱13.4 氯胺T溶液10g/L 临用前称取1.0g氯胺TC7H7ClNNaO2S3H2OchloramineT溶于水并稀释至100mL摇匀贮存于棕色瓶中13.5 异烟酸吡唑啉酮溶液13.5.1 异烟酸溶液称取1.5g异烟酸C6H6NO2isonicotinic acid溶于24mL 2氢氧化钠溶液13.1中加水稀释至100mL 13.5.2 吡唑啉酮溶液称取0.25g吡唑啉酮3甲基1苯基5吡唑啉酮C10H10ON23-methy-1-phenyl-5-pyrazolone溶于20mL NN-二甲基甲酰胺HCONCH32NN-dimethyl formamide 临用前将吡唑啉酮溶液13.5.2和异烟酸溶液13.5.1按15混合13.6 氰化钾KCN标准溶液13.6.1 氰化钾贮备溶液的配制和标定称取0.25g氰化钾KCN注意剧毒溶于1氢氧化钠3.2中并稀释至100mL摇匀避光贮存于棕色瓶中吸取10.00mL上述氰化钾贮备溶液于锥形瓶9.2中加入50mL水和1mL2氢氧化钠13.1加入0.2mL试银灵指示剂8.1用硝酸银标准溶液8.4滴定溶液由黄色刚变为橙红色为止记录硝酸银标准溶液用量V1同时另取10.00mL实验用水代替氰化钾贮备液作空白试验记录硝酸银标准溶液用量V0 氰化物含量c3mg/mL以氰离子CN-计按式3计算300.1004.52013…………………………………………×??VVcc 式中c 硝酸银标准溶液浓度mol/L V1滴定氰化钾贮备溶液时硝酸银标准溶液用量mL V0空白试验硝酸银标准溶液用量mL 52.04相当于1L的1mol/L硝酸银标准溶液的氰离子2CN-的质量g 10.00氰化钾贮备液体积mL 13.6.2 氰化钾标准中间溶液1mL含10.00ìg氰离子先按式4计算出配制500mL1.00mL含10.0ìg氰离子氰化钾标准中间溶液时应吸取氰化钾贮备溶液13.6.1的体积VmL4100050000.10…………………………………………××TV 式中T10001mL氰化钾贮备溶液含氰离子质量ìg 10.001mL氰化钾标准中间溶液含10.00ìg氰离子500氰化钾标准中间溶液体积mL 准确吸取VmL氰化钾贮备溶液13.6.1于500mL棕色容量瓶中用氢氧化钠溶液13.2稀释至标线摇匀13.6.3 氰化钾标准使用溶液1.00mL含1.00ìg氰离子 6 临用前吸取10.00mL氰化钾标准中间溶液13.6.2于100mL棕色容量瓶中用氢氧化钠溶液13.2稀释至标线摇匀仪器14.1 分光光度计或比色计14.2 25mL 具塞比色管操作步骤15.1 校准15.1.l 取8支具塞比色管14.2分别加入氰比钾标准使用溶液13.6.300.200.501.002.003.004.00和5.00mL各加氢氧化钠溶液13.2至10mL 15.1.2 向各管中加入5mL磷酸盐缓冲溶液13.3混匀迅速加入0.2mL氯胺T溶液13.4立即盖塞子混匀放置35min 15.1.3 向各管中加入5mL异烟酸吡唑啉酮溶液13.5混匀加水稀释至标线摇匀在2535的水浴中放置40min 15.1.4 用分光光度计在638nm 波长下用10mm比色皿以试剂空白零浓度作参比测定吸光度并绘制校准曲线15.2 测定15.2.1 分别吸取10.00mL馏出液A6.1和10.00mL空白试验馏出液B6.2于具塞比色管14.2中按15.1.215.1.3和15.1.4进行操作15.2.2 从校准曲线上查出相应的氰化物含量结果计算16.1 计算方法总氰化物含量c4mg/L以氰离子CN-计按式5计算5214……………………………………VVVmmcba 式中ma从校准曲线上查出试份比色时所取馏出液A的氰化物含量ìg mb从校准曲线上查出空白试验馏出液B 的氰化物含量ìg V 样品的体积mL V1试样馏出液A的体积mL V2试份比色时所取馏出液A的体积mL 16.2 精密度和准确度6个实验室测定氰化物含量0.0220.032mg/L 加标水样的结果和测定氰化物含量0.2060.236mg/L加标水样的结果如下1982年10月16.2.1 重复性相对标准偏差分别为7.4和1.8 16.2.2 准确度回收率为9297 第四篇吡啶巴比妥酸比色法原理在中性条件下氰离子和氯胺T的活性氯反应生成氯化氰氯化氰与吡啶反应生成戊烯二醛glutacomdialdehyde戊烯二醛与两个巴比妥酸分子缩合生成红紫色染料进行比色测定试剂718.1 13盐酸HCl 18.2 吡啶巴比妥酸溶液临用前称取0.18g巴比妥酸C4H4N2O37barbituric acid加入3mL吡啶C5H5Npyridine及10mL盐酸18.1待溶解后加水至100mL摇匀贮存在棕色瓶中注本溶液若有不溶物可过滤存于暗处可稳定一天存放于冰箱内可稳定一周18.3 磷酸盐缓冲溶液pH7 称取2.79g无水磷酸二氢钾KH2PO4和4.14g无水磷酸氢二钠Na2HPO4溶于水中稀释至1000mL放入试剂瓶存放冰箱18.4 盐酸溶液0.5mol/L 18.5 酚酞指示剂1 g/L 仪器19.1 分光光度计或比色计19.2 25mL具塞比色管操作步骤20.1 校准20.1.1 取8支具塞比色管19.2分别加入氰化钾标准使用溶液13.6.300.200.501.002.003.004.00和5.00mL各加氢氧化钠13.2至10mL 20.1.2 向各管中加入1滴酚酞指示剂18.5用盐酸18.4调节溶液红色刚消失为止20.1.3 加入5mL磷酸盐缓冲溶液摇匀迅速加入0.2mL氯胺T溶液13.4立即盖塞子混匀放置35min再加入5mL吡啶巴比妥酸溶液18.2加水稀释至标线混匀20.1.4 在40水浴中放置20min取出冷却至室温在分光光度计上在580nm波长处用10mm比色皿以试剂空白零浓度作参比测定吸光度并绘制校准曲线20.2 测定20.2.1 分别取10.00mL馏出液A6.1和10.00mL空白试验馏出液B6.2于具塞比色管19.2中按20.1.220.1.3和21.1.4进行操作20.2.2 从校准曲线上查出相应的氰化物含量结果计算21.1 计算方法总氰化物含量c5mg/L以氰离子CN-计按式6计算6215…………………………………………VVVmmcba 式中ma以校准曲线上查出试份比色时所取馏出液A的氰化物含量ìg mb从校准曲线上查出空白试验馏出液B 的氰化物含量ìg V 样品的体积mL V1试样馏出液A的体积mL V2试份比色时所取馏出液A的体积mL 21.2 精密度和准确度4个实验室测定含氰离子0.0200.024mg/L加标水样结果和0.1480.153mg/L加标水样结果如下1982年10月21.2.1 重复性相对标准偏差分别为4.9和1.5 21.2.2 再现性4个实验室测定0.040mg/L统一已知氰化物样品相对标准偏差为1.2 2l.2.3 准确度4个实验室测定0.040mg/L统一已知氰化物样品相对误差为0.3 8 22 参考文献GB7486-87 附录参考件 A.1 方法中使用计量仪器如天平砝码容量瓶移液管滴定管等应校正后使用A.2 方法中使用的水除特别说明外均用蒸馏水或去离子水A.3 方法中所使用的化学试剂均为分析纯以上试剂 A.4 方法中液体试剂配制的水溶液以试剂的体积加水的体积表示时例如l3盐酸溶液系指1体积盐酸和3体积水混合配制A.5 空白试验系指不加试样以蒸馏水代替试样操作步骤同试样测定的试验。
挥发酚和总氰化物在三种水质中测定方法的比对挥发酚和总氰化物是水质中常见的污染物之一,对人体健康和环境造成严重危害。
准确测定水体中的挥发酚和总氰化物含量对于环境保护和人们的饮用水安全具有重要意义。
本文将对挥发酚和总氰化物在三种不同水质中测定方法进行比对,以探讨其适用性和准确度。
一、挥发酚和总氰化物的测定方法1. 挥发酚的测定方法挥发酚是一类易挥发的有机物,主要来源于石油化工和煤气化工等工业过程以及燃料和溶剂的使用中。
在水体中含有挥发酚会给人体健康和环境造成严重危害。
准确测定水体中挥发酚的含量是非常重要的。
传统的挥发酚测定方法主要有气相色谱法(GC)和高效液相色谱法(HPLC)。
气相色谱法通常用于测定水体中的挥发性有机物,其原理是将挥发性有机物从水体中抽提出来,通过气相色谱仪进行分离和测定。
高效液相色谱法是利用不同分子在液相色谱柱中的分配系数不同来实现分离和测定有机物的方法。
2. 总氰化物的测定方法总氰化物是一类具有高度毒性的无机物,主要来源于金属冶炼、化肥生产和农药生产过程中的废水中。
总氰化物存在于水体中会对水生生物和人体健康造成危害,因此对水体中总氰化物的测定也是非常重要的。
常用的总氰化物测定方法主要有化学法和仪器法。
化学法是通过氰化物离子和铁离子的配位反应生成蓝色络合物,根据络合物的吸光度来测定总氰化物的含量。
仪器法主要是利用离子色谱仪、光度计和原子吸收光谱仪等仪器测定样品中氰化物的含量,具有快速、准确的特点。
二、三种水质的选择为了比对挥发酚和总氰化物的测定方法,在本研究中选择了三种不同水质的样品进行测定,分别为自来水、河水和工业废水。
这三种水质具有不同的特点和来源,可以更好地体现测定方法的适用性和准确度。
1. 自来水自来水是经过处理的饮用水,通常来源于自然水源经过过滤、消毒等处理后供应给人们使用。
自来水的主要污染物为挥发性有机物和重金属等,测定自来水中挥发酚和总氰化物的含量可以评估自来水的安全性。
两种氰化物测定方法的验证与对比摘要:通过对水中氰化物的测定方法《水质(总)氰化物的测定异烟酸-巴比妥酸分光光度法》(HJ 484-2009)与《连续流动注射仪法SXSHJ/ZB002-2015》进行方法验证实验,并对两种方法从标准曲线、样品加标回收率、方法检出限、实验精密度与测定结果、实验试剂消耗与作业速率五个方面对比,得出连续流动注射分析法:具有更良好的线性(r=0.9994~0.9999),较低的检出限(0.00078),回收率为(97.3~101%),相对标准偏差为(1.06~1.26%),相对误差为(0.0010~0.0058)。
较传统方法准确度与可信度更高,作业速率更快,样品与实验试剂消耗量更少,实验对人体的危害更小,适用现代环境监测的定时定量危害小的要求,值得推广应用。
关键词:氰化物;流动分析法;方法验证;方法比对氰化物特指含有氰基的化合物,有剧毒。
水中氰化物通常产生于电镀、冶炼、化学工业等企业的工业废水污染。
氰化物对水环境危害巨大,氰根含量浓度0.01mg/L为浮游生物和甲壳类生物的最大允许浓度,氰根含量浓度为0.04~0.1mg/L升时,就可以使鱼类致死。
此外,水中的氰化物还会使得农业减产、牲畜发病或死亡。
因此,在对氰化物检测中获得的数据的准确性与可靠性就显得尤为重要。
本文通过对传统的分光光度法与自编的连续流动法分别做方法验证,并在方法验证实验的过程中对两种方法进行比对。
1、实验1.1、总氰化物的测定方法1.1.1、异烟酸-巴比妥酸分光光度法(1)、异烟酸-巴比妥酸分光光度法是国内测定总氰化物常用的手工方法,稳定且可靠,该方法基本原理是,首先对样品水样进行化学蒸馏方法预处理,随后在弱酸性条件下,将水样中氰化物与氯胺T发生反应形成氯化氰,然后与异烟酸反应,而后经水解生成戊烯二醛,最后再与巴比妥酸反应得到紫蓝色物质,用比色皿在波长为600nm处测量其吸光度。
1.1.2、连续流动分析法(1)、连续流动法是我中心测定总氰化物常用的方法,作用机理是在酸碱度pH为3.8的环境下,络合氰化物在紫外消解器的作用下消解。
总氰化物GB7486--87 总氰化物是指在磷酸和EDTA存在下,pH小于2的介质中,加热蒸馏,能形成氰化氢的氰化物,包括全部简单氰化物(多为碱金属和碱土金属的氰化物,铵的氰化物)和绝大部分络合氰化物(锌氰络合物、铁氰络合物、镍氰络合物、铜氰络合物等),不包括钴氰络合物。
预处理1.方法原理向水样中加入磷酸和Na2-EDTA,在pH<2条件下,,加热蒸馏,利用金属离子与EDTA络合能力比与氰离子络合能力强的特点,使络合氰化物离解出氰离子,并以氰化氢形式被蒸馏出来,并用氢氧化钠溶液吸收。
仪器(1) 500ml全玻璃蒸馏器。
(2) 600W或800W可调电炉。
(3) 100ml量筒或容量瓶。
(4) 仪器装置。
试剂(1) 磷酸:ρ=1.69g/m1。
(2) 1%(m/V)氢氧化钠溶液。
(3) 10%(m/V)Na2—EDTA溶液。
(4) 乙酸铅试纸:称取5g三水合乙酸铅溶于水中,稀释到100ml。
将滤纸条浸入上述溶液中,1h后,取出晾干,盛于广口瓶中,密塞保存。
(5) 碘化钾—淀粉试纸:称取1.5g可溶性淀粉,用少量水搅成糊状,加入200ml沸水,混匀。
放冷,加0.5g碘化钾和0.5g碳酸钠,用水稀释到250ml,将滤纸条浸渍后,取出晾干,盛于棕色瓶中密塞保存。
(6) l十5硫酸溶液。
(7) 1.26%(m/V)亚硫酸钠溶液。
(8) 氨基磺酸。
(9) 4%(m/V)氢氧化钠溶液。
步骤1.氰化氢的释放和吸收(1) 量取200m1样品,移入500m1蒸馏瓶中(若氰化物含量高,可酌量少取,加水稀释至200ml),加数粒玻璃珠。
(2) 往接收容器内,加入10m1 1%氢氧化钠溶液,作为吸收液。
(3) 馏出液导管上端接冷凝管的出口,下端插入接收容器的吸收液中,检查连接部位,使其严密。
(4) 将10ml Na2—EDTA溶液加入蒸馏瓶内。
(5) 迅速加入10m1磷酸,当样品碱度大时,可适当多加磷酸,使pH<2;立即塞好瓶塞。
总氰化物GB7486--87 总氰化物是指在磷酸和EDTA存在下,pH小于2的介质中,加热蒸馏,能形成氰化氢的氰化物,包括全部简单氰化物(多为碱金属和碱土金属的氰化物,铵的氰化物)和绝大部分络合氰化物(锌氰络合物、铁氰络合物、镍氰络合物、铜氰络合物等),不包括钴氰络合物。
预处理1.方法原理向水样中加入磷酸和Na2-EDTA,在pH<2条件下,,加热蒸馏,利用金属离子与EDTA络合能力比与氰离子络合能力强的特点,使络合氰化物离解出氰离子,并以氰化氢形式被蒸馏出来,并用氢氧化钠溶液吸收。
仪器(1) 500ml全玻璃蒸馏器。
(2) 600W或800W可调电炉。
(3) 100ml量筒或容量瓶。
(4) 仪器装置。
试剂(1) 磷酸:ρ=1.69g/m1。
(2) 1%(m/V)氢氧化钠溶液。
(3) 10%(m/V)Na2—EDTA溶液。
(4) 乙酸铅试纸:称取5g三水合乙酸铅溶于水中,稀释到100ml。
将滤纸条浸入上述溶液中,1h后,取出晾干,盛于广口瓶中,密塞保存。
(5) 碘化钾—淀粉试纸:称取1.5g可溶性淀粉,用少量水搅成糊状,加入200ml沸水,混匀。
放冷,加0.5g碘化钾和0.5g碳酸钠,用水稀释到250ml,将滤纸条浸渍后,取出晾干,盛于棕色瓶中密塞保存。
(6) l十5硫酸溶液。
(7) 1.26%(m/V)亚硫酸钠溶液。
(8) 氨基磺酸。
(9) 4%(m/V)氢氧化钠溶液。
步骤1.氰化氢的释放和吸收(1) 量取200m1样品,移入500m1蒸馏瓶中(若氰化物含量高,可酌量少取,加水稀释至200ml),加数粒玻璃珠。
(2) 往接收容器内,加入10m1 1%氢氧化钠溶液,作为吸收液。
(3) 馏出液导管上端接冷凝管的出口,下端插入接收容器的吸收液中,检查连接部位,使其严密。
(4) 将10ml Na2—EDTA溶液加入蒸馏瓶内。
(5) 迅速加入10m1磷酸,当样品碱度大时,可适当多加磷酸,使pH<2;立即塞好瓶塞。
打开冷凝水,调节可调电炉,由低档逐渐升高,以2—4m1/min的馏出液速度进行加热蒸馏。
(6) 接收瓶内溶液近100m1时,停止蒸馏,用少量水洗馏出液导管,取出接收瓶,用水稀释至标线。
此碱性馏出液(C),供测定总氰化物用。
2.空白试验用实验用水代替样品,按步骤(1)-- (6)操作,得到空白试验馏出液(D),供测定总氰化物用。
硝酸银滴定法概述1.方法原理经蒸馏得到的碱性馏出液(A),用硝酸银标准溶液滴定,氰离子与硝酸银作用形成可溶性的银氰络合离子[Ag(CN)2]¯,过量的银离子与试银灵指示液反应,溶液由黄色变为橙红色,即为终点。
2.方法的适用范围当水样中氰化物含量在1mg/L以上时,可用硝酸银滴定法进行测定。
检测上限为l00mg/L。
本方法适用于饮用水、地面水、生活污水和工业废水。
仪器(1) 10m1棕色酸式滴定管。
(2) 120ml柄皿或150m1锥形瓶。
试剂1.试银灵指示液称取0.02g试银灵溶于100m1丙酮中。
贮存棕色瓶暗处,可稳定一个月。
2.铬酸钾指示液称取10g铬酸钾溶于少量水中,滴加硝酸银溶液至产生橙红色沉淀为止,放置过夜后,过滤,用水稀释至100m1。
3.0.0100mo1/L氯化钠标准溶液称取氯化钠(经600℃干燥lh,在干燥器内冷却)0.5844g置于烧杯中,用水溶解,移入1000m1容量瓶,并稀释至标线,混合摇匀。
4.0.0100mo1/L硝酸银标准溶液(1) 称取1.699g硝酸银溶于水中,稀释至1000m1,贮于棕色试剂瓶中,摇匀,待标定后使用。
(2) 硝酸银溶液的标定:吸取0.0100mo1/L氯化钠标准溶液10.00m1,于150ml柄皿或锥形瓶中,加50m1水。
同时另取一柄皿或锥形瓶,加入60ml水作空白试验。
向溶液中加入3—5滴铬酸钾指示液,在不断搅拌下,从滴定管加入待标定的硝酸银溶液直至溶液由黄色变成浅砖红色为止,记下读数(V)。
同样滴定空白溶液,读数为(V 0),按下式计算:硝酸银标准溶液浓度(mo1/L) = 00.10V V c -⨯ 式中, c ——氯化钠标准溶液浓度(mo1/L);V ——滴定氯化钠标准溶液时,硝酸银溶液用量(m1);V 0——滴定空白溶液时,硝酸银溶液用量(m1)。
5.氢氧化钠溶液配制2% (m /V)的氢氧化钠溶液。
步 骤1.样品测定取100ml 馏出液A (如试样中氰化物含量高时,可酌量少取,用水稀释至100ml)于柄皿或锥形瓶中。
加入0.2m1试银灵指示液,摇匀。
用硝酸银标准溶液滴定至溶液由黄色变为橙红色止,记下读数( V a )。
2.空白试验另取100m1空白试验馏出液B ,于锥形瓶,按样品测定(2)进行滴定,记下读数(V 0)。
计 算氰化物(CN¯, mg/L) =()V V V V V c a 100004.52210⨯⨯⨯-式中,c ——硝酸银标准溶液浓度(mol/L);V a——测定试样时,硝酸银标准溶液用量(ml);V0——空白试验时,硝酸银标准溶液用量(m1);V——样品体积(m1);V1——试样(馏出液A)的体积(ml);V2——试样(测定时,所取馏出液A)的体积(m1);52.04——氰离子(2CN¯)摩尔质量(g/mo1)。
注意事项用硝酸银标准溶液滴定试样前,应以pH试纸试验试样的pH值。
必要时,应加氢氧化钠溶液调节pH>11。
异烟酸—吡唑啉酮光度法概述1.方法原理在中性条件下,样品中的氰化物与氯胺T反应生成氯化氰,再与异烟酸作用,经水解后生成戊烯二醛,最后与吡唑啉酮缩合生成蓝色染料,其色度与氰化物的含量成正比,进行光度测定。
2.方法的适用范围异烟酸—吡唑啉酮比色法,最低检测浓度为0.004mg/L;测定上限为0.25mg/L。
本方法适用于饮用水、地面水、生活污水和工业废水。
仪器(1) 分光光度计或光度计(2) 25ml具塞比色管。
试剂(1) 2%(m/V)氢氧化钠溶液。
(2) 0.1%(m/V)氢氧化钠溶液。
(3) 磷酸盐缓冲溶液(pH=7):称取34.0g无水磷酸二氢钾和35.5g无水磷酸氢二钠于烧杯内,加水溶解后,稀释至1000m1,摇匀。
于冰箱中保存。
(4) 1%(m/V)氯胺T溶液:临用前,称取0.5g氯胺T溶于水,并稀释至50ml,摇匀。
贮存棕色瓶中。
(5) 异烟酸—吡唑啉酮溶液:①异烟酸溶液:称取1.5g异烟酸溶于24m1 2%氢氧化钠溶液中,加水稀释至100ml。
②吡唑啉酮溶液:称取0.25g吡唑啉酮溶于20m1 N, N--二甲基甲酰胺。
临用前,将吡唑啉酮溶液②和异烟酸溶液①按1+5混合均匀。
(6)氰化钾标准溶液:称取0.25g氰化钾(注意剧毒!)溶于0.1%氢氧化钠溶液中,并用0.1%氢氧化钠溶液至100m1,摇匀。
避光贮存于棕色瓶中。
吸取10.00ml氰化钾贮备溶液于锥形瓶中,加入50ml水和1ml 2%氢氧化钠溶液,加入0.2ml 试银灵指示液,用硝酸银标准溶液(0.0100mol/L )滴定,溶液由黄色刚变为橙红色止,记录硝酸银标准溶液用量(V 1)。
同时另取10ml 实验用水代替氰化钾贮备液作空白试验,记录硝酸银标准溶液用量(V 0),按下式计算:氰化物(mg/ml )=()00.1004.5201⨯-V V c 式中,c--硝酸银标准溶液浓度(mol/L );V 1--滴定氰化钾贮备液时,硝酸银标准溶液用量(ml );V 0--空白试验,硝酸银标准溶液用量(ml );52.04—氰离子(2CN¯)的摩尔质量(g/mol );10.00—取用氰化钾贮备液体积(ml )。
(7)氰化钾标准中间溶液(1ml 含10.00µg 氰离子):先按下式计算出配制500ml 氰化钾标准中间液所需氰化钾贮备溶液的体积(V ): V = 10050000.10⨯⨯T 式中,10.00—1ml 氰化钾标准中间溶液含10.00µg CN¯;500—氰化钾标准中间溶液体积(ml );T —氰化钾贮备液含CN¯数(mg )。
准确吸取氰化钾贮备液于500ml 棕色容量瓶中,用0.1%氢氧化钠溶液稀释到标线,摇匀。
(8)氰化钾标准使用溶液(1ml 含1.00µg 氰离子):临用前,吸取10.00ml 氰化钾标准中间溶液(1ml 含10.00µg 氰离子)于100ml 棕色容量瓶中,用0.1%氢氧化钠溶液稀释到标线,摇匀。
步 骤1. 校准曲线的绘制(1) 取8支25ml 具塞比色管,分别加入氰化钾标准使用溶液0、0.20、0.50、1.00、2.00、3.00、4.00、5.00,各加0.1%氢氧化钠溶液到10ml 。
(2) 向各管中加入5m1磷酸盐缓冲溶液,混匀。
迅速加入0.2m1氯胺T 溶液,立即盖塞子,混匀,放置3—5 min 。
(3)向管中加入5m1异烟酸—吡唑啉酮溶液,混匀。
加水稀释至标线,摇匀。
在25--35℃的水浴中放置40min 。
(4) 用分光光度计,在638nm 波长下,用10mm 比色皿,零浓度空白管作参比,测量吸光度,并绘制校准曲线。
2. 样品的测定(1)分别吸取10.00ml 馏出液A 和10.00ml 空白试验馏出液于具塞比色管中,然后,按校准曲线的绘制步骤(2)--(4)进行操作,测量吸光度。
(2)从校准曲线上查出相应的氰化物含量。
计 算氰化物(CN¯,mg/L )=21V V V m m b a •-式中,m a --从校准曲线上查出试样的氰化物含量(µg);m b--从校准曲线上查出空白试样(馏出液B)的氰化物含量(µg);V——样品的体积(ml);V1--试样(馏出液A)的体积(ml);V2--试样(比色时,所取馏出液A)的体积(ml)。
精密度和准确度用加标水样,其氰化物含量为0.022--0.032mg/L,经6个实验室分析,得单个实验室相对标准偏差分别为7.4%和1.8%,加标回收率为92--99%。
注意事项(1)当氰化物以HCN存在时,易挥发。
因此,从加缓冲液后,每一步骤都要迅速操作,并随时盖严塞子。
(2)为降低试验空白值,实验中以选用无色的N-2甲基甲酰胺为宜。
(3)实验温度低时,磷酸盐缓冲溶液会析出结晶,而改变溶液的pH值。
因此,需要在水浴中使结晶溶解,混匀后,方可使用。
(4)当吸收液用较高浓度的氢氧化钠溶液时,加缓冲液前应以酚酞为指示剂,滴加盐酸溶液至红色褪去。
水样和校准曲线均应为相同的氢氧化钠浓度。
吡啶一巴比妥酸光度法概述1.方法原理在中性条件下,氰离子和氯胺T反应生成氯化氰,氯化氰与吡啶反应生成戊烯二醛,戊烯二醛与两个巴比妥酸分子结合生成红紫色染料,进行光度测定。
2.方法的适用范围吡啶-巴比妥酸光度法的最低检测浓度为0.002mg/L,检测上限为0.45mg/L(1cm比色皿)。