第三章 序列两两比对
- 格式:ppt
- 大小:409.50 KB
- 文档页数:51
实验三:两条序列比对与多序列比对实验目的:学会使用MegAlign,ClustalX和MUSCLE进行两条序列和多条序列比对分析实验内容:双序列比对是使两条序列产生最高相似性得分的序列排列方式和空格插入方式。
两条序列比对是生物信息学最基础的研究手段。
第一次实验我们用dotplot方法直观地认识了两条序列比对。
但是dotplot仅仅是展示了两条序列中所有可能的配对,并不是真正意义上的序列比对。
这里介绍进行两条序列比对的软件-MegAlign。
多序列比对是将多条序列同时比对,使尽可能多的相同(或相似)字符出现在同一列中。
多序列比对的目标是发现多条序列的共性。
如果说序列两两比对主要用于建立两条序列的同源关系,从而推测它们的结构和功能,那么,同时比对多条序列对于研究分子结构、功能及进化关系更为有用。
多序列比对对于系统发育分析、蛋白质家族成员鉴定、蛋白质结构预测、保守模块的搜寻等具有非常重要的作用。
我们这节课主要学习多条序列比对的软件-ClustalX, MUSCLE。
一、MegAlignDNASTAR公司的Lasergene软件包是一个比较全面的生物信息学软件,它包含了7个模块。
其中MegAlign可进行两条或多条序列比对分析。
1. 两条序列比对1.1 安装程序解压DNASTAR Lasergene软件压缩包,双击Lasergene710WinInstall.exe文件,按照默认路径安装软件到自己电脑上。
1.2 载入序列a.点击开始-程序-Lasergene-MegAlign,打开软件。
我们首先用演示序列(demo sequence)学习软件的使用。
演示序列所在位置:C:\Program files\ DNASTAR\ Lasergene\ Demo Megalign\ Histone Sequences\。
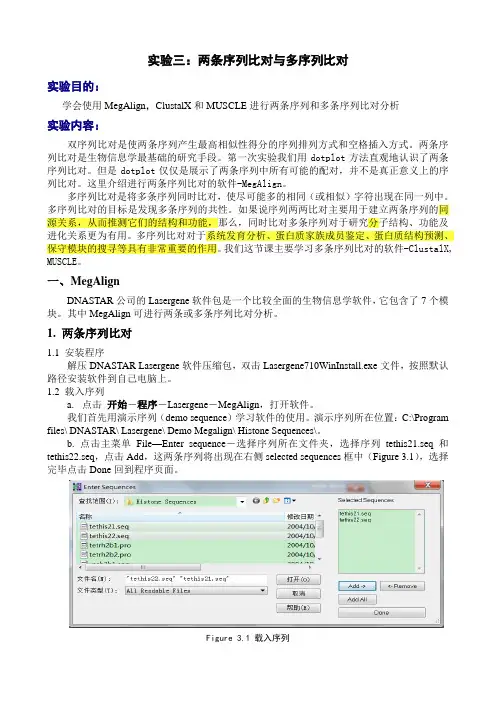
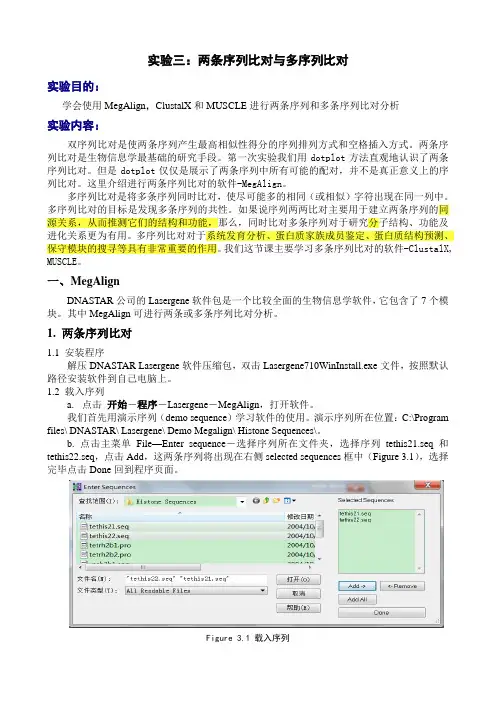
b. 点击主菜单File—Enter sequence-选择序列所在文件夹,选择序列tethis21.seq和tethis22.seq,点击Add,这两条序列将出现在右侧selected sequences框中(Figure 3.1),选择完毕点击Done回到程序页面。




第三章序列比较第二节序列两两比对算法1. 序列两两比对基本算法直接方法—生成两个序列所有可能的比对,分别计算得分(或代价)函数,然后挑选一个得分最高(或代价最小)的比对作代价)函数然后挑选个为最终结果。
本质问题:优化动态规划算法(Dynamic Programming)依据动态规划寻优策略→Needleman-Wunsch算法Needleman Wunsch最短路径问题C1起点终点W1C2W2C1+1路径1:C1 + w1 ?路径2:C2 + w2 ?取最小值!算法求解:从起点到终点逐层计算利用动态规划方法求解序列的两两比对起点终点A ATTC………CGAAG AGTC GAAGGT AGTC………GAAGG ATTC………CGAAG A 1AGTC………GAAGG +T()AATTC CGAAG AATTC………CGAAG AGTC………GAAGG +-T (2)ATTC………CGAAG +A (3)TAGTC………GAAGG-求解过程AATTC………CGAAG起点终点TAGTC………GAAGG•从两个序列前端开始•逐步推进•直到两个序列的末端。
中间过程:比对:S:i与0:T:j序列S: i-1 i i+1序列t: j-1 j j+1序列S: i-1 i i+1 Case1:匹配(si ,tj)序列t: j-1 j j+1序列S: i-1 i i+1序列t: j-1 j j+1序列S: i-1 i i+1Case2:序列t: j-1 j j+1删除(s i ,-)序列S: i-1 i i+1序列t: j-1 j j+1序列S: i-1 i i+1Case3:序列t: j-1 j j+1插入(-,t j )s t m n 。
设序列、的长度分别为和考虑两个前缀0:s:i0:t:jt 所有较短子序列的最优比对即已知假如已知序列0:s:i 和0:t:j 所有较短子序列的最优比对,即已知:(()()1)0:s:(i-1)和0:t:(j-1)的最优比对(2)0:s:(i-1)和0:t:j 的最优比对(3):s:和:t:(j-1)的最优比对0i 0(j 1)则0:s:i 和0:t:j 的最优比对一定是上述三种情况之一的扩展((这取决于(1)替换(s i ,t j )或匹配(s i ,t j ),这取决于s i 是否等于t j ;(2)删除(s i ,-);(3)插入(-,t j )。



实验三:两条序列比对与多序列比对实验目的:学会使用MegAlign,ClustalX和MUSCLE进行两条序列和多条序列比对分析实验内容:双序列比对是使两条序列产生最高相似性得分的序列排列方式和空格插入方式。
两条序列比对是生物信息学最基础的研究手段。
第一次实验我们用dotplot方法直观地认识了两条序列比对。
但是dotplot仅仅是展示了两条序列中所有可能的配对,并不是真正意义上的序列比对。
这里介绍进行两条序列比对的软件-MegAlign。
多序列比对是将多条序列同时比对,使尽可能多的相同(或相似)字符出现在同一列中。
多序列比对的目标是发现多条序列的共性。
如果说序列两两比对主要用于建立两条序列的同源关系,从而推测它们的结构和功能,那么,同时比对多条序列对于研究分子结构、功能及进化关系更为有用。
多序列比对对于系统发育分析、蛋白质家族成员鉴定、蛋白质结构预测、保守模块的搜寻等具有非常重要的作用。
我们这节课主要学习多条序列比对的软件-ClustalX, MUSCLE。
一、MegAlignDNASTAR公司的Lasergene软件包是一个比较全面的生物信息学软件,它包含了7个模块。
其中MegAlign可进行两条或多条序列比对分析。
1. 两条序列比对1.1 安装程序解压DNASTAR Lasergene软件压缩包,双击Lasergene710WinInstall.exe文件,按照默认路径安装软件到自己电脑上。
1.2 载入序列a.点击开始-程序-Lasergene-MegAlign,打开软件。
我们首先用演示序列(demo sequence)学习软件的使用。
演示序列所在位置:C:\Program files\ DNASTAR\ Lasergene\ Demo Megalign\ Histone Sequences\。
b. 点击主菜单File—Enter sequence-选择序列所在文件夹,选择序列tethis21.seq和tethis22.seq,点击Add,这两条序列将出现在右侧selected sequences框中(Figure 3.1),选择完毕点击Done回到程序页面。

第二章:序列的采集和存储2. 序列数据的存储核酸序列数据库国际三大核酸序列数据库:GenBank, EBML, DDBJdbEST: Expressed Sequences Tags数据库UniGene等RefSeq: The Reference Sequence Database蛋白质序列数据库UniProtSwiss—prot & TrEMBL, PIR基因组数据库: Ensembl第三章序列比对I序列间比对的对应关系:匹配、替代、缺失、插入双序列比对算法:Dot matrix(点阵法)动态规划算法Needleman-Wunsch算法Sij = max of Si—1,j-1 + σ(xi , yj )Si—1,j —d ( 从左到右)Si,j—1 —d ( 从上到下)Smith-Waterman 算法Sij = max of 0Si-1,j-1 + σ(xi , yj )Si—1,j -d (从左到右)Si,j—1 -d (从上到下)FASTA和BLAST算法PSI-BLAST (位点特异性迭代BLAST):1. 使用普通的blast算法进行搜索;2。
将搜索得到的序列,包括输入的序列放在一起,构建位点特异性的矩阵(Position Specific Matrix);3。
利用上面得到的矩阵谱(profile),再次在数据库中进行搜索;4. 重复2 ,3 步,直到不再有新的序列出现;PHI—BLAST : 模式发现迭代BLAST第三章序列比对Ⅱ打分矩阵及其含义1,计分方法2, PAM系列矩阵3, BLOSUM 系列矩阵多序列比对:方法改进1。
渐进方法:代表:ClustalW/X, T—Coffee(1)ClustalW/X:计算过程1。
将所有序列两两比对,计算距离矩阵;2. 构建邻接进化树(neighbor—joining tree)/指导树(guide tree);3。
将距离最近的两条序列用动态规划的算法进行比对;4。

第三章序列比较3.3 序列多重比对与序列两两比对不一样,序列多重比对(Multiple Alignment)的目标是发现多条序列的共性。
如果说序列两两比对主要用于建立两条序列的同源关系和推测它们的结构、功能,那么,同时比对一组序列对于研究分子结构、功能及进化关系更为有用。
例如,某些在生物学上有重要意义的相似性只能通过将多个序列对比排列起来才能识别。
同样,只有在多序列比对之后,才能发现与结构域或功能相关的保守序列片段。
对于一系列同源蛋白质,人们希望研究隐含在蛋白质序列中的系统发育的关系,以便更好地理解这些蛋白质的进化。
在实际研究中,生物学家并不是仅仅分析单个蛋白质,而是更着重于研究蛋白质之间的关系,研究一个家族中的相关蛋白质,研究相关蛋白质序列中的保守区域,进而分析蛋白质的结构和功能。
序列两两比对往往不能满足这样的需要,难以发现多个序列的共性,必须同时比对多条同源序列。
图3.14是从多条免疫球蛋白序列中提取的8个片段的多重比对。
这8个片段的多重比对揭示了保守的残基(一个是来自于二硫桥的半胱氨酸,另一个是色氨酸)、保守区域(特别是前4个片段末端的Q-PG)和其他更复杂的模式,如1位和3位的疏水残基。
实际上,多重序列比对在蛋白质结构的预测中非常有用。
多重比对也能用来推测各个序列的进化历史。
从图3.14可以看出,前4条序列与后4条序列可能是从两个不同祖先演化而来,而这两个祖先又是由一个最原始的祖先演化得到。
实际上,其中的4个片段是从免疫球蛋白的可变区域取出的,而另4个片段则从免疫球蛋白的恒定区域取出。
当然,如果要详细研究进化关系,还必须取更长的序列进行比对分析。
对于多重序列比对的定义,实际上是两个序列的推广。
设有k个序列s1, s2, ... ,s k,每个序列由同一个字母表中的字符组成,k大于2;通过插入操作,使得各序列s1, s2, ... ,s k的长度一样,从而形成这些序列的多重比对。
如果将各序列在垂直方向排列起来,则可以根据每一列观察各序列中字符的对应关系,如图3.14。