New protocol for lentiviral vector mass production.
- 格式:pdf
- 大小:839.98 KB
- 文档页数:14

第 44卷第6期2023 年11月Vol.44 No.6November 2023中山大学学报(医学科学版)JOURNAL OF SUN YAT‐SEN UNIVERSITY(MEDICAL SCIENCES)卵巢癌腹腔转移工具细胞的构建及其应用万里舟1,杜汝沛2,陈康梅3(1. 中山大学孙逸仙纪念医院基础与转化医学研究中心,广东广州 510120; 2. 华南理工大学生物医学科学与工程学院,广东广州 511442; 3. 中山大学孙逸仙纪念医院检验科,广东广州 510120)摘要:【目的】 构建稳定共表达荧光素酶(Luc)和增强型绿色荧光蛋白(EGFP)的人源卵巢癌细胞SKOV3(SK-Luc-EGFP)并探究其在卵巢癌腹腔转移的体内外研究中的应用。
【方法】 利用重叠PCR扩增得到的Luc-T2A-EGFP基因片段,构建重组质粒pCDH-Luc-T2A-EGFP-Puro;采用三质粒慢病毒包装系统转染HEK 293T细胞,收集病毒液感染SKOV3细胞;通过嘌呤霉素筛选,流式细胞术检测,筛选高效表达EGFP的细胞(SK-Luc-EGFP),并通过体外生物发光实验验证Luc的表达。
对SK-Luc-EGFP细胞进行如下应用的探究:利用流式细胞术和激光共聚焦区分SK-Luc-EGFP细胞与腹水微环境中的非肿瘤细胞,利用荧光显微镜观察SK-Luc-EGFP与间皮细胞的黏附,利用小动物活体成像仪观察SK-Luc-EGFP细胞的大网膜黏附以及体内成瘤的过程。
【结果】 成功构建慢病毒载体pCDH-Luc-T2A-EGFP-Puro;感染并筛选得到SK-Luc-EGFP单克隆细胞株。
通过荧光显微镜和流式细胞术均能检测到EGFP的表达,细胞纯度达100%;通过流式细胞术和激光共聚焦成像可直观辨别SK-Luc-EGFP细胞与腹水微环境细胞。
体外生物发光试验成功验证Luc的表达,且细胞数和生物发光信号的线性相关系数为0.997 9。

pLKO.1 – TRC Cloning VectorAddgene Plasmid 10878. Protocol Version 1.0. December 2006.Copyright Addgene 2006, All Rights Reserved. This protocol is provided for your convenience. See ―warranty information‖ in ap pendix.Table of Contents∙ A. pLKO.1-TRC Cloning Vector∙ A.1 The RNAi Consortium∙ A.2 Map of pLKO.1∙ A.3 Related plasmids∙ B. Designing shRNA Oligos for pLKO.1∙ B.1 Determine the optimal 21-mer targets in your gene∙ B.2 Order oligos compatible with pLKO.1∙ C. Cloning shRNA oligos into pLKO.1∙ C.1 Recommended materials∙ C.2 Annealing oligos∙ C.3 Digesting pLKO.1 TRC-Cloning Vector∙ C.4 Ligating and transforming into bacteria∙ D. Screening for Inserts∙ D.1 Recommended materials∙ D.2 Screening for inserts∙ E. Producing Lentiviral Particles∙ E.1 Recommended materials∙ E.2 Protocol for producing lentiviral particles∙ F. Infecting Target Cells∙ F.1 Recommended materials∙ F.2 Determining the optimal puromycin concentration∙ F.3 Protocol for lentiviral infection and selection∙G. Safety∙H. References∙H.1 Published articles∙H.2 Web resources∙I. Appendix∙I.1 Sequence of pLKO.1 TRC-Cloning Vector∙I.2 Recipes∙I.3 Warranty informationBack to TopA. pLKO.1-TRC Cloning VectorA.1 The RNAi ConsortiumThe pLKO.1 cloning vector is the backbone upon which The RNAi Consortium has built a library of shRNAs directed against 15,000 human and 15,000 mouse genes. Addgene is working with the TRC to make this shRNA cloning vector available to the scientific community. Please cite Moffat et al., Cell 2006 Mar; 124(6):1283-98 ('PubMed‖:/pubmed/16564017?dopt=abstract) in all publications arising from the use of this vector.A.2 Map of pLKO.1pLKO.1 is a replication-incompetent lentiviral vector chosen by the TRC for expression of shRNAs. pLKO.1 can be introduced into cells via direct transfection, or can be converted into lentiviral particles for subsequent infection of a target cell line. Once introduced, the puromycin resistance marker encoded in pLKO.1 allows for convenient stable selection.Figure 1 : Map of pLKO.1 containing an shRNA insert. The original pLKO.1-TRC cloning vector has a 1.9kb stuffer that is released by digestion with AgeI and EcoRI. shRNA oligos are cloned into the AgeI and EcoRI sites in place of the stuffer. The AgeI site is destroyed in most cases(depending on the target sequence), while the EcoRI site is preserved. For a complete map of pLKO.1 containing the 1.9kb stuffer, visit /10878.Description Vector ElementHuman U6 promoter drives RNA Polymerase III transcription for generation of shRNAU6transcripts.Central polypurine tract, cPPT, improves transduction efficiency by facilitating nuclearcPPTimport of the vector’s preintegration complex in the transduced cells.hPGK Human phosphoglycerate kinase promoter drives expression of puromycin.Puro R Puromycin resistance gene for selection of pLKO.1 plasmid in mammalian cells.sin 3’LTR 3’ Self-inactivating long terminal repeat.f1 ori f1 bacterial origin of replication.Amp R Ampicillin resistance gene for selection of pLKO.1 plasmid in bacterial cellspUC ori pUC bacterial origin of replication.5’LTR 5’ long terminal repeat.RRE Rev response element.A.3 Related ProductsThe following plasmids available from Addgene are recommended for use in conjunction with the pLKO.1 TRC-cloning vector.Plasmid (Addgene ID #) DescriptionpLKO.1 – TRC control Negative control vector containing non-hairpin insert.pLKO.1 – scramble shRNA Negative control vector containing scrambled shRNA.psPAX2Packaging plasmid for producing viral particles.pMD2.G Envelope plasmid for producing viral particles.Note: pLKO.1 can also be used with packaging plasmid pCMV-dR8.2 dvpr and envelope plasmid pCMV-VSVG from Robert Weinberg’s lab. For more information, visit Addgene’s Mammalian RNAi Tools page.Several other laboratories have deposited pLKO derived vectors that may also be useful for your experiment. To see these vectors, visit Addgene’s website and ―search for ―pLKO‖―.Back to TopB. Designing shRNA Oligos for pLKO.1B.1 Determining the Optimal 21-mer Targets in your GeneSelection of suitable 21-mer targets in your gene is the first step toward efficient gene silencing. Methods for target selection are continuously being improved. Below are suggestions for target selection.1. Use an siRNA selection tool to determine a set of top-scoring targets for your gene. For example, the Whitehead Institute for BiomedicalResearch hosts an siRNA Selection Program that can be accessed after a free registration (/bioc/siRNAext/). If you have MacOS X, another excellent program is iRNAi, which is provided free by the company Mekentosj (/irnai/).A summary of guidelines for designing siRNAs with effective gene silencing is included here:∙Starting at 25nt downstream of the start codon (ATG), search for 21nt sequences that match the pattern AA(N 19 ). If no suitable match is found, search for NAR(N 17 )YNN, where N is any nucleotide, R is a purine (A,G), and Y is a pyrimidine (C,U).∙G-C content should be 36-52%.∙Sense 3’ end should have low stability – at least one A or T between position 15-19.∙Avoid targeting introns.∙Avoid stretches of 4 or more nucleotide repeats, especially repeated Ts because polyT is a termination signal for RNA polymerase III.2. To minimize degradation of off-target mRNAs, use NCBI’s BLAST program. Select sequences that have at least 3 nucleotide mismatches to allunrelated genes.Addgene recommends that you select multiple target sequences for each gene. Some sequences will be more effective than others. In addition, demonstrating that two different shRNAs that target the same gene can produce the same phenotype will alleviate concerns about off-target effects.B.2 Ordering Oligos Compatible with pLKO.1To generate oligos for cloning into pLKO.1, insert your sense and antisense sequences from step B.1 into the oligos below. Do not change the ends; these bases are important for cloning the oligos into the pLKO.1 TRC-cloning vector.Forward oligo:5’ CCGG—21bp sense—CTCGAG—21bp antisense—TTTTTG 3’Reverse oligo:5’ AAT TCAAAAA—21bp sense—CTCGAG—21bp antisense 3’For example, if the target sequence is (AA)TGCCTACGTTAAGCTATAC, the oligos would be:Forward oligo:5’ CCGG AATGCCTACGTTAAGCTATAC CTCGAG GTATAGCTTAACGTAGGCATT TTTTTG 3’Reverse oligo:5’ AATTCAAAAA AATGCCTACGTTAAGCTATAC CTCGAG GTATAGCTTAACGTAGGCATT3’Back to TopC. Cloning Oligos into pLKO.1The pLKO.1-TRC cloning vector contains a 1.9kb stuffer that is released upon digestion with EcoRI and AgeI.The oligos from section B contain the shRNA sequence flanked by sequences that are compatible with the sticky ends of EcoRI and AgeI. Forward and reverse oligos are annealed and ligated into the pLKO.1 vector, producing a final plasmid that expresses the shRNA of interest.C.1 Recommended MaterialsMaterial Vendor and catalog #AgeI New England Biolabs (NEB) #R0552SEcoRI NEB #R0101ST4 DNA ligase NEB #M0202SNEB buffer 2 NEB #B7002SDH5 alpha competent cells Invitrogen #18258-012Qiaquick gel extraction kit Qiagen #28704Low melting point agarose Sigma #A9414Luria Broth Agar (LB agar) American Bioanalytical: #AB01200-02000Ampicillin VWR: #7177-48-2. Use at 100 μg/mL.Carbenicillin VWR: #80030-956. Use at 100 μg/mL.C.2 Annealing Oligos1. Resuspend oligos in ddH2O to a concentration of 20 μM, then mix:5 μL Forward oligo5 μL Reverse oligo5 μL 10x NEB buffer 235 μL ddH2O2. Incubate for 4 minutes at 95°C in a PCR machine or in a beaker of boiling water.3. If using a PCR machine, incubate the sample at 70°C for 10 minutes then slowly cool to room temperature over the period of several hours. If using a beaker of water, remove the beaker from the flame, and allow the water to cool to room temperature. This will take a few hours, but it is important for the cooling to occur slowly for the oligos to anneal.C.3 Digesting pLKO.1 TRC Cloning Vector1. Digest pLKO.1 TRC-cloning vector with AgeI. Mix:6 μg pLKO.1 TRC-cloning vector (maxiprep or miniprep DNA)5 μL 10x NEB buffer 11 μL AgeIto 50 μL ddH2O> Incubate at 37°C for 2 hours.2. Purify with Qiaquick gel extraction kit. Elute in 30 μL of ddH2O.3. Digest eluate with EcoRI. Mix:30 μL pLKO.1 TRC-cloning vector digested with AgeI5 μL 10x NEB buffer for EcoRI1 μL EcoRI14 μL ddH2O> Incubate at 37°C for 2 hours.4. Run digested DNA on 0.8% low melting point agarose gel until you can distinctly see 2 bands, one 7kb and one 1.9kb. Cut out the 7kb band and place in a sterile microcentrifuge tube.When visualizing DNA fragments to be used for ligation, use only long-wavelength UV light. Short wavelength UV light will increase the chance of damaging the DNA.5. Purify the DNA using a Qiaquick gel extraction kit. Elute in 30 μL of ddH2O.6. Measure the DNA concentration.C.4 Ligating and Transforming into Bacteria1. Use your ligation method of choice. For a standard T4 ligation, mix:2 μL annealed oligo from step C.2.20 ng digested pLKO.1 TRC-cloning vector from step C.3. (If you were unable to measure the DNA concentration, use 1 μL)2 μL 10x NEB T4 DNA ligase buffer1 μL NEB T4 DNA ligaseto 20 μL ddH2O> Incubate at 16°C for 4-20 hours.2. Transform 2 μL of ligation mix into 25 μL competent DH5 alpha cells, following manufacturer’s protocol. Plate on LB agar plates containing 100 μg/mL ampicillin or carbenicillin (an ampicillin analog).Back to TopD. Screening for InsertsYou may screen for plasmids that were successfully ligated by restriction enzyme digestion. However, once you have identified the positive clones, it is important to verify the insert by conducting a sequencing reaction.D.1 Recommended MaterialsMaterial Vendor and catalog #DNA Miniprep Kit Qiagen #27104EcoRI NEB #R0101SNcoI NEB #R0193SAgarose Sigma #A9539D.2 Screening for InsertsDay 1:1. Innoculate 5 colonies from each ligation into LB + 100 μg/mL ampicillin or carbenicillin.Day 2:2. Spin down the cultures and use a miniprep kit to obtain DNA.3. Conduct a restriction digest with EcoRI and NcoI:∙ 1 μg miniprep DNA∙ 2 μL 10x NEB buffer for EcoRI∙0.8 μL EcoRI∙0.8 μL NcoI∙to 20 μL ddH2O> Incubate at 37°C for 1-2 hours.4. Run the digestion products on a 1% agarose gel. You should see two fragments, a 2kb fragment and a 5kb fragment.5. Sequence positive clones with pLKO.1 sequencin g primer (5’ CAA GGC TGT TAG AGA GAT AAT TGG A 3’).You may need to adjust the sequencing conditions if the DNA polymerase has difficulty reading through the secondary structure of the hairpin sequence.Back to TopE. Producing Lentiviral ParticlesBefore this step, you must contact your institution’s Bio-Safety office to receive permission and institution-specific instructions. You must follow safety procedures and work in an environment (e.g. BL2+) suitable for handling HIV-derivative viruses.For transient knockdown of protein expression, you may transfect plasmid DNA directly into the target cells. The shRNA will be expressed, but the DNA is unlikely to be integrated into the host genome.For stable loss-of-function experiments, Addgene recommends that you generate lentiviral particles and infect the target cells. Addition of puromycin will allow you to select for cells that stably express your shRNA of interest.E.1 Recommended MaterialsMaterial Vendor and catalog #psPAX2Addgene #12260pMD2.G Addgene #12259HEK-293T cells GenHunter: #Q401FuGENE® 6 Transfection Reagent Roche Applied Biosciences: #11814443001OPTI-MEM® serum-free media Invitrogen: #31985Dulbecco’s Modified Eagle Medium (DMEM) Invitrogen: #11995Fetal Bovine Serum (FBS) Invitrogen: #16000Penicillin/Streptomycin Invitrogen: #15140-122Polypropylene tubes VWR: #87003-290Note: pLKO.1 could also be packaged using pCMV-dR8.2 dvpr and pCMV-VSVG from the Robert Weinberg lab. For more information, visit Addgene’s Mammalian RNAi Tools page.E.2 Protocol for Producing Lentiviral ParticlesThis protocol is for transfection in a 6 cm plate. The protocol can be scaled to produce different amounts of virus as needed.Day 1:a. For each plasmid to be transfected, plate 7×105 HEK-293T cells in 5 mL of media in a 6 cm tissue culture plate. Incubate cells at 37oC, 5%CO2 overnight.Although cells should regularly be passaged in DMEM + 10% FBS with penicillin/streptomycin, cells should be plated at this step in DMEM + 10% FBS without antibiotics (no penicillin or streptomycin).Day 2:b. Perform the transfection in the late afternoon because the transfection mix should only be incubated with the cells for 12-15 hours.c. In polypropylene microfuge tubes (do NOT use polystyrene tubes), make a cocktail for each transfection:∙ 1 μg pLKO.1 shRNA plasmid∙750 ng psPAX2 packaging plasmid∙250 ng pMD2.G envelope plasmid∙to 20 μl serum-free OPTI-MEMYou may want to vary the ratio of shRNA plasmid, packaging plasmid, and envelope plasmid to obtain the ratio that gives you the optimal viral production.d. Create a master mix of FuGENE® 6 transfection reagent in serum-free OPTI-MEM. Calculate the amount of Fugene® and OPTI-MEMnecessary given that each reaction will require 6 μL FuGENE® + 74 μL OPTI-MEM. For example:∙1x master mix: 6 μL FuGENE® + 74 μL OPTI-MEM∙5x master mix: 30 μL FuGENE® + 370 μL OPTI-MEM∙10x master mix: 60 μL FuGENE® + 740 μL OPTI-MEMIn a polypropylene tube, add OPTI-MEM first. Pipette FuGENE® directly into the OPTI-MEM – do not allow FuGENE® to come in contact with the walls of the tube before it has been diluted. Mix by swirling or gently flicking the tube. Incubate for 5 minutes at room temperature.e. Add 80 μL of FuGENE® master mix to each tube from step c for a total volume of 100 μL. Pipette master mix directly into the liquid and notonto the walls of the tube. Mix by swirling or gently flicking the tube.f. Incubate for 20-30 minutes at room temperature.g. Retrieve HEK-293T cells from incubator. The cells should be 50-80% confluent and in DMEM that does not contain antibiotics.h. Without touching the sides of the dish, gently add DNA:FuGENE® mix dropwise to cells. Swirl to disperse mixture evenly. Do not pipette orswirl too vigorously, as you do not want to dislodge the cells from the plate.i. Incubate cells at 37°C, 5% CO2 for 12-15 hours.Day 3:j. In the morning, change the media to remove the transfection reagent. Replace with 5 mL fresh DMEM + 10% FBS + penicillin/streptomycin.Pipette the media onto the side of the plate so as not to disturb the transfected cells.k. Incubate cells at 37°C, 5% CO2 for 24 hours.Day 4:l. Harvest media from cells and transfer to a polypropylene storage tube. The media contains your lentiviral particles. Store at 4°C.m. Add 5 mL of fresh media containing antibiotics to the cells and incubate at 37°C, 5% CO2 for 24 hours.Day 5:n. Harvest media from cells and pool with media from Day 4. Spin media at 1,250 rpm for 5 minutes to pellet any HEK-293T cells that were inadvertently collected during harvesting.In lieu of centrifugation, you may filter the media through a 0.45 μm filter to remove the cells. Do not use a 0.2 μm filter, as this is likely to shear the envelope of your virus.o. Virus may be stored at 4°C for a few days, but should be frozen at -20°C or -80°C for long-term storage.Freeze/thaw cycles decrease the efficiency of the virus, so Addgene recommends that you use the virus immediately or aliquot the media into smaller tubes to prevent multiple freeze/thaw cycles.Back to TopF. Infecting Target CellsLentiviral particles can efficiently infect a broad range of cell types, including both dividing and non-dividing cells. Addition of puromycin will allow you to select for cells that are stably expressing your shRNA of interest.F.1. Recommended MaterialsMaterial Vendor and catalog #Hexadimethrine bromide (polybrene)* Sigma-Aldrich: #H9268Protamine Sulfate* MP Biomedicals: #194729Puromycin* Sigma-Aldrich: #P8833Target cells Varies based on your experimentCulture media for target cells Varies based on your experimentMaterials for assay Varies based on your experimentDetailed protocols for preparing polybrene, protamine sulfate, and puromycin are locat ed in the ―Appendix‖.F.2. Determining the Optimal Puromycin ConcentrationEach cell line responds differently to puromycin selection. Addgene strongly recommends that you determine the optimal puromycinconcentration for your cell line before initiating your experiment.Day 1:a. Plate target cells in ten 6 cm plates and grow at 37° C, 5% CO2 overnight.Day 2:b. The target cells should be approximately 80-90% confluent.c. Dilute puromycin in the preferred culture media for your target cells. The final concentration of puromycin should be from 1-10 μg/mL in 1μg/mL increments.d. Label plates from 1-10 and add appropriate puromycin-containing media to cells.Days 3+:e. Examine cells each day and change to fresh puromycin-containing media every other day.f. The minimum concentration of puromycin that results in complete cell death after 3-5 days is the concentration that should be used forselection in your experiments. (You may wish to repeat this titration with finer increments of puromycin to determine a more precise optimal puromycin concentration.)F.3. Protocol for Lentiviral Infection and SelectionDay 1:a. Plate target cells and incubate at 37°C, 5% CO2 overnight.Day 2:b. Target cells should be approximately 70% confluent. Change to fresh cul ture media containing 8 μg/mL polybrene.Polybrene increases the efficiency of viral infection. However, polybrene is toxic to some cell lines. In these cell lines, substitute protamine sulfate for polybrene.c. Add lentiviral particle solution from step E. For a 6 cm target plate, add between 0.05-1 mL virus (add ≥0.5 mL for a high MOI, and ≤0.1 mLfor a low MOI). Scale the amount of virus added depending on the size of your target plate.MOI (multiplicity of infection) refers to the number of infecting viral particles per cell. Addgene recommends that you test a range of MOIs to determine the optimal MOI for infection and gene silencing in your target cell line.d. Incubate cells at 37°C, 5% CO2 overnight.Day 3:e. Change to fresh media 24 hours after infection.If viral toxicity is observed in your cell line, you may decrease the infection time to between 4 – 20 hours. Remove the virus-containing media and replace with fresh media. Do not add puromycin until at least 24 hours after infection to allow for sufficient expression of the puromycin resistance gene.f. To select for infected cells, add puromycin to the media at the concentration determined in step E.2.Addgene recommends that you maintain one uninfected plate of cells in parallel. This plate will serve as a positive control for the puromycin selection.Days 4+:g. Change to fresh puromycin-containing media as needed every few days.h. Assay infected cells. The following recommendations are guidelines for the number of days you should wait until harvesting your cells.However, you should optimize the time based on your cell line and assay:Assay Days post-infectionmRNA knockdown (quantitative PCR) ≥ 3 daysProtein knockdown (western blot) ≥ 4 daysPhenotypic assay ≥ 4 daysBack to TopG. SafetyBL2 safety practices should be followed when preparing and handling lentiviral particles. Personal protective clothing should be worn at all times. Use plastic pipettes in place of glass pipettes or needles. Liquid waste should be decontaminated with at least 10% bleach. Laboratory materials that come in contact with viral particles should be treated as biohazardous waste and autoclaved. Please follow all safety guidelines from your institution and from the CDC and NIH for work in a BL2 facility.If you hav e any questions about what safety practice to follow, please contact your institution’s safety office.To obtain the MSDS for this product, visit /sitemap and follow the MSDS link.Back to TopH. ReferencesH.1. Published ArticlesKhvorova A et. al. 2003. Functional siRNAs and miRNAs exhibit strand bias. Cell 115:209-216. (PubMed)Moffat J et. al. 2006. A lentiviral RNAi library for human and mouse genes applied to an arrayed viral high-content screen. Cell 124:1283-1298.(PubMed)Naldini L et. al. 1996. In vivo gene delivery and stable transduction of nondividing cells by a lentiviral vector. Science 272:263-267. (PubMed)Schwarz DS et. al. 2003. Asymmetry in the assembly of the RNAi enzyme complex. Cell 115:199-208. (PubMed)Stewart SA et. al. 2003. Lentivirus-delivered stable gene silencing by RNAi in primary cells. RNA 9(4):493-501. (PubMed)Zufferey R et. al. 1997. Multiply attenuated lentiviral vector achieves efficient gene delivery in vivo. Nat Biotechnol 15(9):871-5. (PubMed)Zufferey R et. al. 1998. Self-inactivating lentivirus vector for safe and efficient in vivo gene delivery. J Virol 72(12):9873-80. (PubMed)H.2. Web resourcesAddgene’s mammalian RNAi web site: /mammalianrnaiThe RNAi Consortium (TRC): /genome_bio/trc/rnai.htmlBackground on RNAi mechanism: /focus/rnai/animations/animation/animation.htmWhitehead siRNA Selection Program: /bioc/siRNAext/Mekentosj iRNAi Program: /irnai/Back to TopI. AppendixI.1. Sequence of pLKO.1 TRC-Cloning VectorClick here to see the sequence of pLKO.1 TRC-cloning vector. The vector is 8901 base pairs total, and the stuffer insert is shown in all capital letters.I.2. RecipesLuria Broth Agar (LB agar) + antibioticPer 40 grams of powder from American Bioanalytical catalog # AB01200-02000, LB contains:10g tryptone5g yeast extract10g sodium chloride15g agar> Prepare LB agar solution by dissolving 40g of LB powder in 1L of distilled water. Autoclave and cool to 55°C. Add 1mL of 100mg/mL ampicillin or carbenicillin to obtain a final concentration of 100 μg/mL antibiotic. Pour plates and store at 4°C.Hexadimethrine Bromide (Polybrene)Prepare a 1mg/mL solution of polybrene (Sigma-Aldrich catalog #H9268) in 0.9% NaCl. Autoclave to sterilize. Stock solution is stable at 4°C for up to one year. The powder form of polybrene is stable at 4°C for several years.Protamine SulfateStore protamine sulfate (MP Biomedicals catalog #194729) at 4°C. Freely soluble in hot water and slightly soluble in cold water.PuromycinPrepare a 50mg/mL stock solution of puromycin (Sigma-Aldrich catalog #P8833) in distilled water. Sterilize by passing through a 0.22 μm filter.Store aliquots at -20°C.I.3. Warranty InformationAddgene is committed to providing scientists with high-quality goods and services. Addgene makes every effort to ensure the accuracy of its literature, but realizes that typographical or other errors may occur. Addgene makes no warranty of any kind regarding the contents of any literature. Literature are provided t o you as a guide and on an ―AS IS‖ ―AS AVAILABLE‖ basis without warranty of any kind either expressed or implied, including but not limited to the implied warranties of fitness for a particular purpose, non-infringement, typicality, safety and accuracy.The distribution of any literature by Addgene is not meant to carry with it, and does not grant any license or rights of access or use to the materials described in the literature.The distribution of materials by Addgene is not meant to carry with it, and does not grant any license, express or implied, under any patent. All transfers of materials from Addgene to any party are governed by Addgene’s Terms of Use, Addgene’s Terms of Purchase, and applicable Material Transfer Agreements between the party that deposited the material at Addgene and the party receiving the material.Back to Top。


Generation of gene-engineered hematopoietic stem cellsCliniMACS Prodigy® Hematopoietic Stem Cell Engineering SystemApplicationThe CliniMACS Prodigy® Hematopoietic Stem Cell Engineering System allows fully automated transduction of human CD34+ cells from patient material for the generation of gene-engineered hematopoietic stem cells (HSCs).This application sheet gives an overview of the process, the specifications, and materials needed. In addition, it elucidates the setup of the CliniMACS Prodigy Tubing Set (TS) 520 and the performance data.SpecificationsCellular starting material:C D34+ cells , e.g., enrichedfrom mobilized leukapheresis Starting cell number:at least 2×107 cellsStarting sample volume:40–250 mLFinal product: gene-engineered CD34+ cells Final product volume:100 mLProcess time:2–3 days* A mount is given for the one-hit lentiviral transduction of 1×108 cellsin 100 mL. Please discuss your specific requirements with yourMiltenyi Biotec representative.1Process overview for HSC engineering* T he duration of the HSC engineering processdepends on the number of transduction cycles 23CliniMACS Prodigy TS 520 setup for HSC engineeringPrinciple of the HSC engineering process on the CliniMACS Prodigy®Miltenyi Biotec B.V. & Co. KG | Phone +49 2204 8306-0 | Fax +49 2204 85197|**********************|Miltenyi Biotec provides products and services worldwide. Visit /local to find your nearest Miltenyi Biotec contact.130-126-605Unless otherwise specifically indicated, Miltenyi Biotec products and services are for research use only and not for therapeutic or diagnostic use. MACS® GMP Products are for research use and ex vivo cell culture processing only, and are not intended for human in vivo applications. For regulatory status in the USA, please contact your local representative. MACS GMP Products are manufactured and tested under a quality system certified to ISO 13485 and are in compliance with relevant GMP guidelines. They are designed following the recommendations of USP <1043> on ancillary materials. The CliniMACS® System components, including Reagents, Tubing Sets, Instruments, and PBS/EDTA Buffer, are designed, manufactured and tested under a quality system certified to ISO 13485.In the EU, the CliniMACS System components are available as CE-marked medical devices for their respective intended use, unless otherwise stated. The CliniMACS Reagents and Biotin Conjugates are intended for in vitro use only and are not designated for therapeutic use or direct infusion into patients. The CliniMACS Reagents in combination with the CliniMACS System are intended to separate human cells. Miltenyi Biotec as the manufacturer of the CliniMACS System does not give any recommendations regarding the use of separated cells for therapeutic purposes and does not make any claims regarding a clinical benefit. For the manufacturing and use of target cells in humans the national legislation and regulations – e.g. for the EU the Directive 2004/23/EC (“human tissues and cells”), or the Directive 2002/98/EC (“human blood and blood components”) – must be followed. Thus, any clinical application of the target cells is exclusively within the responsibility of the user of a CliniMACS System.In the US, the CliniMACS CD34 Reagent System, including the CliniMACS Plus Instrument, CliniMACS CD34 Reagent, CliniMACS Tubing Sets TS and LS, and the CliniMACS PBS/EDTA Buffer, is FDA approved as a Humanitarian Use Device (HUD), authorized by U.S. Federal law for use in the treatment of patients with acute myeloid leukemia (AML) in first complete remission. The effectiveness of the device for this indication has not been demonstrated. All other products of the CliniMACS Product Line are available for use only under an approved Investigational New Drug (IND) application or Investigational Device Exemption (IDE). CliniMACS, CliniMACS Prodigy, MACS, and the MACS logo are registered trademarks or trademarks of Miltenyi Biotec and/or its affiliates in various countries worldwide. Copyright © 2020 Miltenyi Biotec and/or its affiliates. All rights reserved.Human CD34+ cells enriched from eight mobilized leukapheresis products were transferred into HSC-Brew GMP Medium supplemented with MACS® GMP Recombinant Human Flt3-Ligand, SCF, TPO, and IL-3 (day 0) and transduced with lentiviral vector encoding GFP (MOI 100) on day 1 using the CliniMACS Prodigy® HSC Engineering System. The same experiment was performed manually following a standard protocol using 24-well plates. (A ) On day 2, similar cell recovery was detected when using the CliniMACS Prodigy HSC Engineering System and manual handling. (B ) Harvested gene-engineered CD34+ cells were further cultured manually in 96-well plates and transduction efficiency was measured on day 5. Transduction efficiency of CD34+ cells processed with the CliniMACS Prodigy was comparable to that of cells processed manually.Performance data。

lentimpra实验流程English Answer:Introduction.Lentimpra is a lentiviral vector platform that enables the efficient and stable delivery of therapeutic genes into target cells. Lentiviral vectors are derived from HIV-1 and have the ability to integrate their genetic material into the host cell genome, providing long-term gene expression. Lentimpra vectors are engineered to be replication-incompetent, minimizing the risk of insertional mutagenesis and other safety concerns.Experimental Workflow.The lentimpra experimental workflow typically involves the following steps:1. Vector Design and Construction: The first step is todesign and construct the lentiviral vector. This involves selecting the appropriate promoter and therapeutic gene, as well as designing the necessary regulatory elements. The vector is then constructed using molecular cloning techniques.2. Vector Production: The next step is to produce the lentiviral vector. This is typically done by transfecting a packaging cell line with the vector plasmid. The packaging cell line produces the viral particles that will carry the therapeutic gene into target cells.3. Vector Purification: Once the lentiviral particles have been produced, they are purified to remove any impurities or contaminants. This is typically done using ultracentrifugation or other purification methods.4. Target Cell Transduction: The purified lentiviral vector is then transduced into target cells. This is typically done by incubating the target cells with the vector in the presence of a transduction reagent.5. Selection and Expansion: After transduction, the target cells are selected for those that have successfully integrated the vector into their genome. This is typically done using a selectable marker gene, such as antibiotic resistance. The selected cells are then expanded to generate a population of genetically modified cells.6. Assessment of Gene Expression: The final step is to assess the expression of the therapeutic gene in the genetically modified cells. This can be done using avariety of methods, such as quantitative PCR, Western blotting, or immunohistochemistry.Applications.Lentimpra has a wide range of applications in gene therapy, including:Cancer immunotherapy: Lentimpra can be used to deliver genes encoding immune stimulatory molecules into cancer cells, enhancing the immune response against the tumor.Gene correction: Lentimpra can be used to deliver genes that correct genetic defects in patients with inherited diseases.Stem cell therapy: Lentimpra can be used to deliver genes into stem cells, enabling their differentiation into specific cell types for use in regenerative medicine.Advantages.Lentimpra offers several advantages over other gene delivery methods:High transduction efficiency: Lentiviral vectors have a high transduction efficiency, allowing for a large proportion of target cells to be genetically modified.Stable gene expression: Lentiviral vectors integrate their genetic material into the host cell genome, providing long-term gene expression.Broad cell tropism: Lentiviral vectors can transduce awide range of cell types, including stem cells, immune cells, and cancer cells.Safety: Lentimpra vectors are engineered to be replication-incompetent, minimizing the risk of insertional mutagenesis and other safety concerns.Conclusion.Lentimpra is a versatile and efficient gene delivery platform that has the potential to revolutionize the field of gene therapy. Its ability to deliver therapeutic genes into target cells with high efficiency and long-term stability makes it an ideal tool for a wide range of applications, including cancer immunotherapy, gene correction, and stem cell therapy.Chinese Answer:概述。
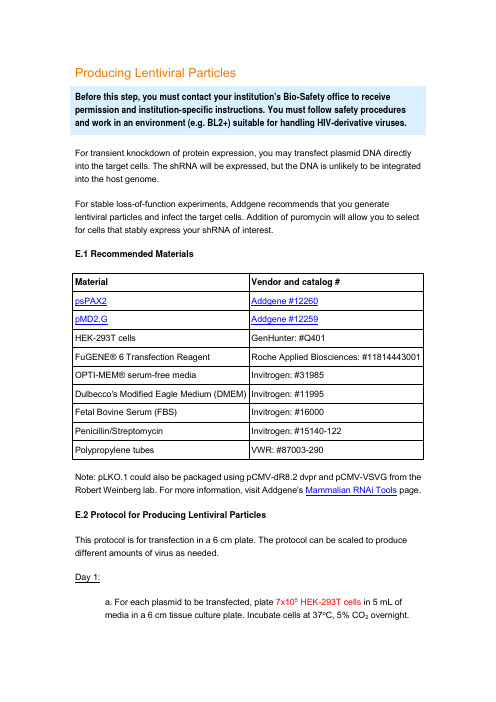
Producing Lentiviral ParticlesBefore this step, you must contact your institution's Bio-Safety office to receive permission and institution-specific instructions. You must follow safety procedures and work in an environment (e.g. BL2+) suitable for handling HIV-derivative viruses.For transient knockdown of protein expression, you may transfect plasmid DNA directly into the target cells. The shRNA will be expressed, but the DNA is unlikely to be integrated into the host genome.For stable loss-of-function experiments, Addgene recommends that you generate lentiviral particles and infect the target cells. Addition of puromycin will allow you to select for cells that stably express your shRNA of interest.E.1 Recommended MaterialsNote: pLKO.1 could also be packaged using pCMV-dR8.2 dvpr and pCMV-VSVG from the Robert Weinberg lab. For more information, visit Addgene's Mammalian RNAi Tools page.E.2 Protocol for Producing Lentiviral ParticlesThis protocol is for transfection in a 6 cm plate. The protocol can be scaled to produce different amounts of virus as needed.Day 1:a. For each plasmid to be transfected, plate 7x105 HEK-293T cells in 5 mL ofmedia in a 6 cm tissue culture plate. Incubate cells at 37o C, 5% CO2 overnight.Although cells should regularly be passaged in DMEM + 10% FBS withpenicillin/streptomycin, cells should be plated at this step in DMEM + 10%FBS without antibiotics (no penicillin or streptomycin).Day 2:b. Perform the transfection in the late afternoon because the transfection mixshould only be incubated with the cells for 12-15 hours.c. In polypropylene microfuge tubes (do NOT use polystyrene tubes), make acocktail for each transfection:1 μg pLKO.1 shRNA plasmid750 ng psPAX2 packaging plasmid250 ng pMD2.G envelope plasmidto 20 μl serum-free OPTI-MEMYou may want to vary the ratio of shRNA plasmid, packaging plasmid, and envelope plasmid to obtain the ratio that gives you the optimal viralproduction.d. Create a master mix of FuGENE® 6 transfection reagent in serum-freeOPTI-MEM. Calculate the amount of Fugene® and OPTI-MEM necessary giventhat each reaction will require 6 μL FuGENE® + 74 μL OPTI-MEM. For example:1x master mix: 6 μL FuGENE® + 74 μL OPTI-MEM5x master mix: 30 μL FuGENE® + 370 μL OPTI-MEM10x master mix: 60 μL FuGENE® + 740 μL OPTI-MEMIn a polypropylene tube, add OPTI-MEM first. Pipette FuGENE® directly into theOPTI-MEM - do not allow FuGENE® to come in contact with the walls of the tube before it has been diluted. Mix by swirling or gently flicking the tube. Incubate for 5 minutes at room temperature.e. Add 80 μL of FuGENE® master mix to each tube from step c for a total volumeof 100 μL. Pipette master mix directly into the liquid and not onto the walls of thetube. Mix by swirling or gently flicking the tube.f. Incubate for 20-30 minutes at room temperature.g. Retrieve HEK-293T cells from incubator. The cells should be 50-80% confluentand in DMEM that does not contain antibiotics.h. Without touching the sides of the dish, gently add DNA:FuGENE® mix dropwiseto cells. Swirl to disperse mixture evenly. Do not pipette or swirl too vigorously, as you do not want to dislodge the cells from the plate.i. Incubate cells at 37o C, 5% CO2 for 12-15 hours.Day 3:j. In the morning, change the media to remove the transfection reagent. Replacewith 5 mL fresh DMEM + 10% FBS + penicillin/streptomycin. Pipette the mediaonto the side of the plate so as not to disturb the transfected cells.k. Incubate cells at 37o C, 5% CO2 for 24 hours.Day 4:l. Harvest media from cells and transfer to a polypropylene storage tube. Themedia contains your lentiviral particles. Store at 4o C.m. Add 5 mL of fresh media containing antibiotics to the cells and incubate at 37o C, 5% CO2 for 24 hours.Day 5:n. Harvest media from cells and pool with media from Day 4. Spin media at 1,250 rpm for 5 minutes to pellet any HEK-293T cells that were inadvertently collectedduring harvesting.In lieu of centrifugation, you may filter the media through a 0.45 μm filter to remove the cells. Do not use a 0.2 μm filter, as this is likely to shearthe envelope of your virus.o. Virus may be stored at 4o C for a few days, but should be frozen at -20o C or-80o C for long-term storage.Freeze/thaw cycles decrease the efficiency of the virus, so Addgene recommends that you use the virus immediately or aliquot the media intosmaller tubes to prevent multiple freeze/thaw cycles.Back to TopF. Infecting Target CellsLentiviral particles can efficiently infect a broad range of cell types, including both dividing and non-dividing cells. Addition of puromycin will allow you to select for cells that are stably expressing your shRNA of interest.F.1. Recommended Materials* Detailed protocols for preparing polybrene, protamine sulfate, and puromycin are located in the Appendix.F.2. Determining the Optimal Puromycin ConcentrationEach cell line responds differently to puromycin selection. Addgene strongly recommends that you determine the optimal puromycin concentration for your cell line before initiating your experiment.Day 1:a. Plate target cells in ten 6 cm plates and grow at 37o C, 5% CO2 overnight.Day 2:b. The target cells should be approximately 80-90% confluent.c. Dilute puromycin in the preferred culture media for your target cells. The finalconcentration of puromycin should be from 1-10 μg/mL in 1 μg/mL increments.d. Label plates from 1-10 and add appropriate puromycin-containing media tocells.Days 3+:e. Examine cells each day and change to fresh puromycin-containing media everyother day.f. The minimum concentration of puromycin that results in complete cell deathafter 3-5 days is the concentration that should be used for selection in yourexperiments. (You may wish to repeat this titration with finer increments ofpuromycin to determine a more precise optimal puromycin concentration.)F.3. Protocol for Lentiviral Infection and SelectionDay 1:a. Plate target cells and incubate at 37o C, 5% CO2 overnight.Day 2:b. Target cells should be approximately 70% confluent. Change to fresh culturemedia containing 8 μg/mL polybrene.Polybrene increases the efficiency of viral infection. However,polybrene is toxic to some cell lines. In these cell lines, substituteprotamine sulfate for polybrene.c. Add lentiviral particle solution from step E. For a 6 cm target plate, add between0.05-1 mL virus(add ≥0.5 mL for a high MOI, and ≤0.1 mL for a low MOI). Scalethe amount of virus added depending on the size of your target plate.MOI (multiplicity of infection) refers to the number of infecting viral particles per cell. Addgene recommends that you test a range of MOIs todetermine the optimal MOI for infection and gene silencing in your targetcell line.d. Incubate cells at 37o C, 5% CO2 overnight.Day 3:e. Change to fresh media 24 hours after infection.If viral toxicity is observed in your cell line, you may decrease the infection time to between 4 - 20 hours. Remove the virus-containing mediaand replace with fresh media. Do not add puromycin until at least 24 hoursafter infection to allow for sufficient expression of the puromycin resistancegene.f. To select for infected cells, add puromycin to the media at the concentrationdetermined in step E.2.Addgene recommends that you maintain one uninfected plate of cells in parallel. This plate will serve as a positive control for the puromycinselection.Days 4+:g. Change to fresh puromycin-containing media as needed every few days.h. Assay infected cells. The following recommendations are guidelines for the number of days you should wait until harvesting your cells. However, you should optimize the time based on your cell line and assay:Assay Days post-infectionmRNA knockdown (quantitative PCR) ≥ 3 daysProtein knockdown (western blot) ≥ 4 daysPhenotypic assay ≥ 4 days。
维特比算法基因组序列全文共四篇示例,供读者参考第一篇示例:维特比算法是一种常用于基因组序列分析的算法,它是一个概率模型,通常用于预测最可能的序列。
在基因组学研究中,通过维特比算法可以有效地识别基因结构和进行基因组序列比对,进而推断基因功能和进行基因突变分析。
基因组序列是生物体内的所有基因的总和,它记录了生物体内所含有的所有遗传信息。
通过对基因组序列的研究,科学家们可以了解生物体的遗传背景,预测基因功能,甚至研究基因突变的影响。
基因组序列的长度通常非常庞大,因此如何高效地分析和处理这些序列成为了研究者们面临的首要挑战。
维特比算法正是为了解决这一难题而被广泛应用的。
它是一种最大后验概率准则下的解码算法,通过动态规划的方式计算出基因组序列中最可能的路径,并输出这条路径对应的序列。
维特比算法的优势在于其高效性和准确性,能够有效地处理大规模的基因组序列数据。
在维特比算法中,首先需要构建一个状态转移矩阵和一个发射概率矩阵。
状态转移矩阵描述了基因组序列中不同状态之间的转移概率,比如嘌呤到嘌呤、嘌呤到嘧啶等。
发射概率矩阵则描述了每个状态发射不同碱基的概率,比如在嘌呤状态下发射A的概率、C的概率等。
通过这两个矩阵,可以计算出基因组序列中每个碱基的概率分布。
接着,维特比算法利用动态规划的思想遍历整个基因组序列,计算出每个位置上最可能的状态。
具体来说,对于每个位置i和每个状态j,维特比算法会计算出到达位置i并处于状态j的最大可能概率。
通过不断更新这些概率值,最终可以得到整条基因组序列中最可能的状态路径。
一旦得到最可能的状态路径,就可以根据状态路径和发射概率矩阵推断出具体的碱基序列。
这个过程可以帮助研究人员快速准确地识别基因结构、预测基因功能和进行基因序列比对。
维特比算法还可以用于研究基因突变的影响,通过比较不同基因组序列间的状态路径,可以发现可能的突变点并推断其影响。
维特比算法在基因组序列分析中起到了至关重要的作用。
通信原理维特比解码Matlab实现维特比解码(Viterbi decoding)是一种广泛应用于通信系统的算法,尤其在卷积码的解码中。
以下是一个基于Matlab的维特比解码实现的简要指南,主要包含初始化参数、构建状态网格、计算路径概率、更新最佳路径、计算最终解码结果、输出解码数据、性能评估和可视化结果等步骤。
1.初始化参数2.在开始维特比解码之前,需要设置一些参数,如卷积码的生成多项式、约束长度、编码比特率等。
这些参数将用于构建状态网格和计算路径概率。
3.构建状态网格4.状态网格是用于表示卷积码状态转移的图。
在Matlab中,可以使用二维数组来表示状态网格。
每个状态由一个二元组表示,例如 (00,01)、(00,10)、(01,00) 等。
5.计算路径概率6.在构建了状态网格之后,需要计算每个状态到接收序列的路径概率。
这可以通过使用动态规划的方法来完成。
在Matlab中,可以使用递归函数来计算路径概率。
7.更新最佳路径8.在计算了所有路径概率之后,需要更新最佳路径。
最佳路径是指具有最大概率的路径。
在Matlab中,可以使用动态规划的思想来更新最佳路径。
9.计算最终解码结果10.一旦找到了最佳路径,就可以根据该路径计算出原始的未编码数据。
在Matlab中,可以使用一个递归函数来解码最佳路径并输出原始数据。
11.输出解码数据12.最后,将解码后的数据输出到控制台或保存到文件中。
在Matlab中,可以使用fprintf函数将数据写入文件,或者使用disp函数将数据输出到控制台。
13.性能评估14.为了评估维特比解码的性能,可以使用一些性能指标,如误码率(BER)和误帧率(FER)。
在Matlab中,可以使用循环和条件语句来计算这些性能指标。
15.可视化结果16.最后,可以将维特比解码的过程进行可视化,以更直观地了解解码性能。
在Matlab中,可以使用plot函数来绘制状态网格和最佳路径等图示。
Vector的操作方法一)对分子序列的操作我们以一个DNA序列为例,进行一系列的常规分析;最后将此DNA序列翻译成氨基酸序列,并对此氨基酸序列进行各种分析A,DNA序列为猪生长激素的cDNA序列,长为761bp。
首先使用Vector NTI的Create New 命令将此序列导入到Vector NTI的数据库中:1,第一种方法:如果只知道序列时,点击Molecule才菜单中的Create New——Using Sequence Editor(DNA/RNA……);2,在出现的“New DNA/RNA Molecule”对话框中,首先在General填入导入序列的名称——PGH;3,在DNA/RNA Molecule活页中,选中Linear DNA,Animal/other Eukaryotes,Replicon Type 中选Chromosome;4,Description中填入:S.Scrofa Growth hormone mRNA;5,在Sequence and Maps中点击“Edit Sequence”按钮,将DNA序列复制后,点“Paste”按钮-点“OK”-确认后就可以完成序列导入。
B,如果是一个从GenBank上下载的序列文件,则:点击“Molecule” 菜单-Open-Molecule files 命令,找到序列文件,在File format中选中GenBank Files;点击OK。
(二)常规操作:当序列导入完成后,在桌面出现三个窗口,上左侧的窗口中显示的是该序列的常规信息,上右侧窗口则以图形的格式展示序列的特征区及酶切图谱等。
下面一个窗口显示的是序列:默认状态下以双链形式出现,也可以更改为单链显示。
1.选择序列区域:在图形区域或序列区域直接拖动鼠标左键,同时在最下端的状态栏中显示出所选区域的范围。
2.删除:选中后直接点击键盘上的Delete键,确认后即可删除。
3.选中序列片段后,点击Edit菜单,用其中的命令可以完成对此片段的剪切、复制、删除、定义为新的特征区和用其它序列来代替等。
第十八章 Genomic Analysis III第十八章 Genomic Analysis ⅢPromoter binding factors analysisÆ分析基因的轉錄因子結合位置:在GenomBench的功能中,使用者可以找出基因的promoter序列,promoter 位於基因的上游,使用者可以先把欲分析的範圍圈選出來:圖18.1 將欲分析的範圍圈選圈選出來的序列可以複製輸出,這一段部分即為該基因的上游區域,一般而言選取的範圍為基因上游3000-5000nt區域。
遺憾的是NTI沒有分析promoter 序列transcription factor binding site的功能,使用者只能把找到的promoter序列放到相關的分析網站進行transcription factor binding site的分析。
Exon/intron structure analysisÆ由NTI主程式分析基因體結構:使用者欲分析的序列必須存放在主程式中的Local Database(圖18.2),建議使用者將欲比對的序列存放於特定的資料夾中,詳細的操作方法請參考序列資料建立的章節。
圖18.2 將欲比對的序列放在Local Database,左方為Local Database內的檔案該資料夾可存放多條序列,程式會把該資料夾中的所有序列進行分析。
Vector NTI 教育訓練手冊接下來在主程式中開啟序列時請先將”限制酶分析”的功能關閉(圖18.3):圖18.3 關閉”限制酶分析”的功能接著選擇Analysis→GenomBench Tools(圖18.4):圖18.4 比對序列可以選擇兩種方式:SIM4或Spidey比對基因序列和基因體序列有兩種方式:SIM4和Spidey,這兩種方法的計算方式有些許的不同,使用SIM4的視窗(圖18.5)如下圖所示:第十八章 Genomic Analysis III 圖18.5使用SIM4,左上方為此序列名稱,中間為比對的基因序列左上方Sequence to analyze的欄位為目前使用者所分析的基因體序列(圖18.6);中間上方Alignment sequences為提供比對的基因序列,請把資料項目選至上述設定的資料夾:圖18.6 選擇Alignment sequence提供的比對資料右上方的Strand欄位可以選擇只針對正股、互補股或者雙股進行分析。
39Maurizio Federico (ed.), Lentivirus Gene Engineering Protocols, Methods in Molecular Biology, vol. 614, DOI 10.1007/978-1-60761-533-0_2, © Humana Press, a part of Springer Science + Business Media, LLC 2010Chapter 2New Protocol for Lentiviral Vector Mass ProductionMaría Mercedes Segura, Alain Garnier, Yves Durocher, Sven Ansorge, and Amine KamenAbstractMultiplasmid transient transfection is the most widely used technique for the generation of lentiviral vectors. However, traditional transient transfection protocols using 293 T adherent cells and calcium phosphate/DNA co-precipitation followed by ultracentrifugation are tedious, time-consuming, and difficult to scale up. This chapter describes a streamlined protocol for the fast mass production of lentiviral vectors and their purification by affinity chromatography. Lentiviral particles are generated by transient transfection of suspension growing HEK 293 cells in serum-free medium using polyethylenimine (PEI) as transfection reagent. Lentiviral vector production is carried out in Erlenmeyer flasks agitated on orbital shakers requiring minimum supplementary laboratory equipment. Alternatively, the method can be easily scaled up to generate larger volumes of vector stocks in bioreactors. Heparin affinity chromatography allows for selective concentration and purification of lentiviral particles in a singlestep directly from vector supernatants. The method is suitable for the production and purification of different vector pseudotypes.Key words: Lentiviral vector production, Lentiviral vector purification, Scaleable processes, Transient transfection, Cell suspension culture, Serum-free medium, ChromatographyLentiviral vectors play a key role as gene delivery vehicles in many fundamental and applied research applications since, unlike most currently existing gene transfer systems, they are able to provide long-term gene expression. Along with the growing interest in lentiviral vectors, comes the need to develop streamlined production and purification procedures for the generation of high-titer vector stocks. A variety of production systems have shown to efficiently generate transduction-competent lentiviral particles, including transient transfection of mammalian cells (1), the use of stable packaging cell lines (2), and more recently, baculovirus1. Introduction40Segura et al.technology (3). However, multiplasmid transient transfection continues to be the most commonly used technique for the generation of lentiviral stocks, since it constitutes a faster, simpler, and more versatile approach. Transient transfection avoids the time-consuming and tedious process of developing stable packaging cell lines or recombinant baculoviruses prior to lentiviral vector production. In addition, it allows for the use of cytotoxic/cytostatic transgenes and/or vector components, which is the case for many HIV-1-derived proteins (4–7) and for the commonly used vesicular stomatitis virus pseudotyping envelope glycoprotein (VSV-G) (8), that otherwise need to be put under tight regulatory control (9). Furthermore, transient transfection production systems permit testing various transgenes of interest and envelope proteins with alternative cell tropisms in a relatively short time (10).Detailed protocols describing the production of lentiviral stocks by multiplasmid transient transfection of human embryonic kidney (HEK) 293 T cells using calcium phosphate/DNA co-precipitation are available (11–13). In these protocols, producer cells are grown as monolayers in 10 or 15 cm culture dishes in the presence of 10% fetal bovine serum (FBS). Following successful transfection, 10–15 mL of supernatant containing ~106–107 infective lentiviral particles per mL (IVP/mL) can be recovered at day 2 and 3 posttransfection. Subsequently, harvested supernatants are pooled, filtered through 0.45-m m membranes, and usually purified through two rounds of ultracentrifugation in order to improve vector potency and purity. A critical parameter for correct calcium phosphate/DNA co-precipitation is the pH. A strict control of the pH of the reagents used for transfection and the percentage of CO 2 in the incubators is required because slight variations in the pH can lead to the formation of a too fine or too coarse precipitate that will adversely affect transfection efficiency (12–14). Transfection efficiency may also be affected by the lot of FBS used for cell culture (13). Another problem frequently encountered using these protocols is the loose attachment of HEK 293 T producer cells to culture dishes (13–15). This greatly complicates the production process, as extreme care needs to be taken to prevent producer cell detachment during wash and medium replacement steps. In some protocols, this problem has been circumvented by precoating culture dishes with poly-l -lysine (12). It is also important to change the culture medium after transfection since calcium phosphate is toxic to the producer cells, which can further contribute to HEK 293 T cell detachment (14). In order to overcome the practical difficulties associated with these protocols, reduce the time and effort required for vector production and purification, and allow method scalability, we have developed and optimized a new protocol for lentiviral vector production (16).41 New Protocol for Lentiviral Vector Mass Production This protocol describes how to produce lentiviral vectors by polyethylenimine (PEI)-mediated transient transfection of HEK 293 cells grown in suspension culture and serum-free conditions. Protocols for the fast titration and chromatography purification of lentiviral vector stocks are also provided. Transient transfection is achieved using PEI, a cationic polymer, that binds DNA resulting in the formation of a compact DNA–PEI complex (polyplex) that can efficiently transfect mammalian cells. The use of PEI has a number of advantages over calcium phosphate/DNA coprecipita-tion. It allows for efficient transfection of HEK 293 cells, both in adherent and suspension cultures while showing minimal cytotoxic effects. It is much easier to use and does not require a tight control of transfection conditions, which are difficult to achieve at large scale (17). PEI-mediated transfection is effective, both in the pres-ence or absence of serum in the culture medium. Other efficient transfection reagents, such as cationic lipids (e.g., lipofectamine TM), are too expensive particularly when considering large-scale vector production. In this protocol, we describe the production of lentiviral vectors by transient transfection using a third generation lenti-viral vector system. This system, specifically designed to minimize the risk of generating replication-competent virus (RCV) (18), consists of four plasmids: two packaging plasmids coding for Gag/Pol and Rev sequences, a VSV-G plasmid, and a transfer vector plasmid coding for the green fluorescent protein (GFP) marker to facilitate determination of transfection efficiencies and viral titers. It is important to mention that the method has been successfully adapted to the production of lentiviral vectors using a different combination and number of plasmids (unpublished results).One key aspect for the success of this protocol is the use of HEK 293 cells adapted to grow in suspension. Production of len-tiviral vectors using adherent cell cultures can only be scaled up by increasing cell attachment surface. This is typically achieved in the laboratory by increasing the number of dishes used for vector production, which results in a tedious and time-consuming vec-tor production process. Scale-up can also be accomplished by using alternative culture devices (e.g., roller bottles, multitray cell factories, or microcarriers in stirred tanks) that provide extended anchorage surface. However, these cell culture systems are com-plex, costly, and only provide a limited scalability. To overcome these problems, a suspension-adapted HEK 293 cell line is used in our laboratory for lentiviral vector production. Suspension-adapted cells can easily be grown at high cell densities in stirred tanks, which can be scaled up to several thousands of liters. The use of suspension growing cells also avoids potential problems associated with loose attachment of HEK 293 cells to culture dishes and the use of trypsin, which further simplifies the pro-duction technique. Lentiviral vector production is carried out in Erlenmeyer flasks agitated on orbital shakers requiring minimum42Segura et al.supplementary laboratory equipment (CO 2 and humidity-resis-tant orbital shaker). The production scale described in this proto-col is small (125 mL shake flasks containing 20 mL of cell culture). The procedure can be easily scaled up in the laboratory to 650 mL cell culture in 2-L shake flasks. Larger volumes of vector superna-tant have been successfully produced in bioreactors (16). The method can also be easily scaled down for high-throughput screening purposes in multidishes.Another important aspect for the success of this protocol is that the producer cell line used grows in serum-free medium. Serum is the main source of contaminants in harvested superna-tants and is the predominant cost factor for large-scale vector pro-duction. Serum supplementation increases the complexity, duration, and cost of downstream processing operations and also raises regulatory concerns due to the risk of introducing adventi-tious agents. In this chapter, we describe a heparin affinity chro-matography procedure that allows for the concentration and purification of lentiviral supernatants produced under serum-free conditions in a single step (16). Unlike traditional ultracentrifu-gation techniques, chromatography enables fast, efficient, and reproducible separation of viral particles and this purification strategy is scalable (19). In addition, heparin affinity chromatog-raphy purification typically results in high recoveries of infective particles because of the mild conditions required for viral vector binding and elution and should be useful for the purification of different vector pseudotypes (20–22).1. Cell culture capabilities and Biosafety Level II containment (see Note 1).2. Human embryonic kidney 293 cell line (HEK 293) adapted to grow in suspension culture and serum-free medium (Biotechnology Research Institute, National Research Council of Canada) (see Note 2).3. Hyclone SFM4 Transfx293TM medium (HyClone Cat. No. SH30860) supplemented with 0.1% Pluronic ® F-68 (Invitrogen Cat. No. 24040-032) or any other serum-free medium that supports HEK 293 suspension cell growth and allows for PEI-based transfection (see Note 3).4. Sterile Dimethyl Sulfoxide (DMSO), cell culture grade.5. Neubauer hemacytometer and exclusion dye (Trypan Blue 0.1% in PBS or Erythrosin B 25 mg/mL in PBS).6. Disposable 125-mL polycarbonate Erlenmeyer flasks (Corning Life Science, Cat. No. CLS431143) (see Note 4).2. Materials2.1. Suspension CellCulture43New Protocol for Lentiviral Vector Mass Production 7. Humidified cell culture incubator at 37°C containing 5% CO 2 in air. 8. CO 2 and humidity resistant orbital shaker (see Note 5). 1. Escherichia coli DH5a strain (Invitrogen Cat. No. 18265-017) and SURE ® strain (Stratagene Cat. No. 200238). 2. CircleGrow agar plates and medium (Qbiogene Cat. No. 3000-122) containing appropriate selective antibiotic (e.g., 100 m g/mL of ampicillin) (Sigma Cat. No. A9518). 3. Maxi/Giga prep plasmid purification kit (Qiagen Cat. No. 12163/12191). 4. Buffer: 10 mM Tris–HCl, 1 mM EDTA (TE buffer), pH 8. 1. Linear PEI, MW 25,000 (Polysciences Cat. No. 23966). Sterile stock solution: 1 mg/mL of PEI in Milli-Q H 2O, pH 7 (see Note 6). 2. Purified plasmids: Self-inactivating lentiviral transfer vector (pCSII-CMV5-GFPq) (9), Env-protein plasmid (pSVCMVin) coding for the VSV-G protein (23) and third generation packaging plasmids (pMDLg/pRRE#54 and pRSV-Rev) coding for Gag-Pol and Rev sequences, respectively (18). 1. Low-pressure liquid chromatography system (FPLC). 2. Fractogel ® EMD heparin (S) gel (Merck) packed into an HR 5/5 column (GE Healthcare) to a final volume of 1 mL (see Note 7). 3. 0.45 m m pore size Acrodisc syringe-mounted filters with HT Tuffryn ® polysulfone membrane (Pall Life Sciences). 4. Buffer A: 20 mM Tris–HCl, pH 7.5 filtered and degassed (see Note 8). 5. Buffer B: 20 mM Tris–HCl, 2 M NaCl, pH 7.5 filtered and degassed. 6. Storage buffer: 150 mM NaCl in Milli-Q H 2O, 20% Ethanol filtered and degassed. 7. Regeneration/sanitization buffer: 2 M NaCl in Milli-Q H 2O, 0.1 M NaOH filtered and degassed (see Note 9). 1. Flow cytrometry capabilities. 2. HEK 293 cells.3. Multidish 12 well, Nunclon™ (Nunc Cat. No. 150628).4. Polybrene stock 8 mg/mL, filter sterilized.5. Phosphate-buffered saline (PBS), pH 7.4.6. Formaldehide stock 16% in PBS. Keep at 4°C.7. FACS tubes.2.2. PlasmidAmplificationand Purification2.3. PEI-MediatedTransient Transfectionof Suspension Cells2.4. Concentrationand Purificationby Heparin AffinityChromatography2.5. Titrationof Lentiviral Vectorswith Suspension Cells44Segura et al.1. The cell line used for lentivirus vector production and titration is the HEK 293 suspension cell line. This cell line grows in serum-free HyClone SFM4 Transfx293TM medium supple-mented with 0.1% Pluronic ® F-68.2. HEK 293 cells are preserved in liquid nitrogen in cryovials. Cells must be frozen while in exponential cell growth. Each vial should contain 1 mL of cell suspension with 5 × 106 and 5 × 107 viable cells/mL in freezing medium (90% serum-free medium and 10% DMSO). Cryopreserved cells are recovered by rapidly thawing the vial in a 37°C water bath. The entire content of the vial is transferred into a 15 mL tube containing 9 mL of prewarmed serum-free medium and centrifuge at 350 × g for 5–10 min. Resuspend cell pellet in 10 mL of medium, transfer cell suspension to a 10 cm dish and incu-bated 24 h in static conditions before transferring into shake flasks. A new vial is thawed every ~3 months.3. Maintain suspension cell cultures in 125 mL shake flasks con-taining 20 mL of cell suspension. Flasks are placed on an orbital shaker platform rotating at 110–120 rpm incubated at 37°C in a humidified atmosphere containing 5% CO 2 in air (Fig. 1). Cell count and viability are measured daily using a Neubauer hemocytometer and an exclusion dye.4. Subculture cells while in exponential growth phase, i.e., when the cell density reaches 1–2 × 106 cells/mL, dilute the suspen-sion cell culture in fresh medium down to 3 × 105 cells/mL (typically 2 or 3 times a week). HEK 293 cell cultures should grow as single-cell suspensions (see Note 10). Healthy HEK 293 cells show viability over 90% at all times, a doubling time of 24 h and can achieve high cell densities (4–5 × 106 viable cells/mL). 1. Transform E. coli competent cells with the different plasmids required for lentiviral vector production by standard methods (see Note 11). Plate on agar medium with antibiotic andincubate overnight at 37°C. 2. Use freshly transformed cell colonies for plasmid amplifica-tion. Grow bacterial cell cultures in medium containing anti-biotic during 24 h (see Note 12).3. Purify plasmids according to the manufacturer’s instructions. Check the plasmid purity and concentration by UV absor-bance at 260 and 280 nm and DNA integrity by agarose gel electrophoresis (see Note 13). Adjust plasmid concentration to ~1 mg/mL of DNA in TE buffer, aliquot and freeze at −20°C.3. Methods3.1. Suspension CellCulture Procedures3.2. PlasmidAmplificationand Purification45New Protocol for Lentiviral Vector Mass Production 1. The day before the experiment, split HEK 293 cells to a density of 5 × 105 cells/mL (see Note 14). Grow cultures overnight and determine cell density and viability. Cell density at transfection should be ~1 × 106 cells/mL and cell cultureviability over 95%. 2. Transfer 18 mL of cell suspension into a new disposable125 mL flask and return the cell culture to the incubator until transfection (see Note 15). Warm-up the culture medium to 25–37°C. Thaw the DNA and the PEI reagent at roomtemperature. 3. Transfer 2 mL of transfectionmedium into a 15 mL steriletube and add a total of 20 m g of DNA with a VSV-G: Gag-Pol: Rev: transfer vectorplasmid mass ratio of 1:1:1:2. This isequivalent to 4 m g of each plasmid, except for the transfer vector, which is added in excess (8 m g). Vortex gently (see Note 16).4. Add 60 m L of stock PEI solution (60 m g). Vortex gently.5. Incubate the transfection mixture for 15 min at room tem-perature to allow PEI–DNA complex formation.6. Transfect cells by adding the whole PEI–DNA mixture to the culture and swirl the flask (see Notes 17 and 18).3.3. PEI-MediatedTransient Transfectionof Suspension Cells(Fig. 2)Fig. 1. Suspension cell cultures. Suspension cell cultures grown in shake flasks and multidishes agitated on an orbital shaker platform placed in the cell culture incubator46Segura et al.7. Incubate flasks in a 37°C incubator containing a humidified atmosphere of 5% CO 2 in air on an orbital shaker platform rotating at 110–120 rpm.8. Harvest supernatants containing infective lentiviral particles daily by centrifugation of the cell suspension in a 50 mL ster-ile tube at 350 × g during 5-10 min. Store the supernatant at 4°C and resuspend the cell pellet in 20 mL of fresh medium. Return the cell suspension to the incubator in the same shake flask and incubate for another 24 h.9. Pool supernatants containing the highest lentiviral vector titers. Under the conditions described herein, we obtain sus-tained high titers from day 3 to 5 posttransfection. Filter the 60 mL of pooled supernatant through 0.45 m m membranes using an Acrodisc syringe-mounted filter. Store vector stocks at −80°C or keep at 4°C overnight for subsequent chroma-tography purification.1. Start FPLC system and rinse with Milli-Q H 2O.2. Install the 1-mL Fractogel ® heparin column and remove the storage buffer with 10 column volumes (CV) of Milli-Q H 2O at 0.3 mL/min.3. Rinse lines A and B with the corresponding buffers and equilibrate the column with 10 CV of binding/wash buffer containing 150 mM NaCl (7.5% buffer B) at 0.5 mL/min (153 cm/h linear flow rate) (see Note 19).4. Thaw lentivirus supernatants using water bath at 37°C (if required).3.4. Concentrationand Purificationby Heparin AffinityChromatography Fig. 2. Lentiviral vector production strategy. The scheme depicts the production of lentiviral vector particles by PEI-mediatedtransient transfection of HEK 293 suspension growing cells using a third generation lentiviral plasmid system47New Protocol for Lentiviral Vector Mass Production 5. Refilter the sample using an Acrodisc syringe-mounted filter with a pore size of 0.45 m m if the supernatants were frozen. Aliquot starting material samples for analyses.6. Monitor the UV absorbance at 280 nm. When a stable baseline is achieved, load lentivirus supernatant.7. Apply a step-wise gradient elution strategy that includes a wash step at 150 mM NaCl (7.5% buffer B, 15 CV) to remove the bulk of contaminating proteins, followed by virus elution at 350 mM NaCl (17.5% buffer B, 13 CV) and a final high-stringency wash step at 1.2 M NaCl (60% buffer B, 6.5 CV) to remove tightly bound contaminants. The process is carried out at room temperature and 0.5 mL/min.8. The virus particles elute in a defined peak at 350 mM NaCl (Fig. 3). Pool virus-containing fractions and aliquot for analyses.9. After each run, re-equilibrate the column with binding buffer (10 CV) at 0.5 mL/min or store the column in storage buffer (10 CV) at 0.3 mL/min.10. Store purified samples at −80°C along with the previously aliquoted samples.1. Count exponentially growing HEK 293 cells using a Neubauer hemacytometer and an exclusion dye. Warm-up the culture medium to 25–37°C.2. Dilute cells in sterile 50 mL tubes to a density of 5.5 × 105 cells/mL with fresh culture medium. Add polybrene directly to this cell suspension to achieve a final concentration of 8 m g/mL.3. Plate cells in 12-well plates (0.9 mL/well) and transduce with 0.1 mL of virus sample (nondiluted or diluted in fresh culture medium).4. Incubate plates at 37°C on the orbital shaker (110–120 rpm) for 48 h at 37°C in a humidified atmosphere containing 5% CO 2 in air.3.5. Titration ofLentiviral Vectors byFlow Cytometry UsingSuspension GrowingTarget Cells(see Note 20)Fig. 3. Typical heparin affinity chromatography elution profile. Clarified lentiviral vector supernatant (35 mL) was loaded onto a 1 mL Fractogel ® EMD Heparin (S) column. The virions are eluted by addition of 350 mM NaCl into the mobile phase using a previously designed elution strategy for the purification of gamma-retroviral particles (20–22)48Segura et al.5. Transfer transduced cell suspensions to 1.5 mL Eppendorftubes, centrifuge at 350 × g for 5 min and resuspend cellpellet in 0.5 mL of PBS. Vortex mildly.6. Fix cells by addition of 0.5 mL of freshly prepared 4%formaldehyde in PBS and incubate samples for 30 min at4°C (see Note 21).7. Transfer the cell suspension to an FACS tube and determinethe percentage of transduced GFP+ cells by flow cytometryanalysis (see Note 22).8. Viral titers are calculated with the following formula:T iter (IVP/mL) = (%GFP+ cells/100) × (Number of cells attime of exposure) × (virus dilution factor)/(sample volume);being the number of cells at time of exposure ~5 × 105 cellsand the sample volume 1 mL.4. Notes1. Lentiviral vectors must be handled under appropriate bio-safety containment by trained personnel using biologicalsafety cabinets and following the guidelines specified by theinstitution, where the experiments are conducted. The natureof the transgene must be taken into account when establish-ing biosafety requirements. The work described herein hasbeen performed in biosafety level-2 laboratories.2. The cell line used for lentivirus vector production and titra-tion is the HEK 293. The cell line has been developed byNRC for serum-free suspension cell culture (US Patent #6,210,922) and deposited at ATCC (CRL-12585). A cGMPmaster cell bank has been manufactured for clinical applica-tions. In the previous work, the HEK 293 EBNA-1 cell line(clone 6E) stably expressing a truncated functional form ofthe EBV nuclear antigen-1 was used giving similar results.These cell lines have been selected due to their ability to growin suspension under serum-free conditions and produce highamounts of recombinant proteins upon transient transfection.Other highly transfectable suspension and serum-free adaptedcell lines could be employed for vector production, althoughvector yields may be lower.3. Suspension culture medium is typically supplemented with0.1% Pluronic®F-68 to avoid shear stress in culture. Notethat some commercially available suspension culture mediacontain anticlumping agents (e.g., dextran sulfate, heparin)than can inhibit PEI-mediated transfection.4. This protocol describes lentiviral vector production at small scaleusing 125 mL shake flasks containing 20 mL of cell culture.This procedure can be easily scaled up to 650 mL cell culture in a 2-L shake flask. The cell culture working volume should range between 16 and 33% of the total shake flask volume to allow adequate culture oxygenation. Larger volumes of vec-tor supernatant can be produced in bioreactors. The process described here has been successfully scaled up from shake flasks to a 3-L bioreactor (16). The method can also be scaled down for high-throughput screening purposes. In this case, 6-well plates with a final cell suspension culture volume of 2 mL are recommended.5. Ensure that the orbital shaker can function under humid con-ditions. Do not stop the orbital shaker while it is inside the humidified incubator for extended period of time as this may cause the shaker to seize.6. PEI can be dissolved by lowering the pH of the solution below2 by drop wise addition of concentrated HCl. Stir until PEI is completely dissolved and then neutralize with 10 M NaOH. Adjust volume to obtain a final concentration of 1 mg/mL of PEI. Filter-sterilize the solution through a 0.22 m m mem-brane. Aliquot and keep stock solutions at −20°C. 7. It should be noted that some chromatographic columns, suchas the HR5/5, are provided with top and bottom filters designed for protein purification purposes. To avoid losses of viral particles trapped on the filter, replace the original filters with a suitable filter (i.e., 10 m m mesh). 8. It is essential that all buffers used for chromatography arefreshly filtered (0.45 m m) and degassed to protect the column. Samples should also be filtered through 0.45 m m membranes before being loaded into the column. 9. It is recommended that after 3–4 purification runs the col-umns are regenerated/ sanitized. This is normally performed by washing with 2–3 column volumes of regeneration/saniti-zation solution followed by an extensive wash with Milli-Q H 2O to bring back the column to neutral pH. Monitor the column effluent pH before beginning the next run. 10. Suspension cells sometimes grow as small clusters. Vigorousvortexing may be required at each subculture step for a num-ber of passages until the cultures grow predominantly as sin-gle-cell suspensions. Extensive clumping affects cell growth and transfection efficiency. 11. To minimize the frequency of homologous recombinationbetween LTR regions in E. coli , it is recommended that lentiviral transfer vector plasmids be propagated using Rec-bacterial strains (e.g., SURE ® strain). 12. We routinely obtained approximately 10 g of cell pellet forevery 0.5 L of bacterial cell culture and routinely recovered ~10 mg of pure plasmid.13. For optimal transfection efficiency, high-quality plasmidstocks should be used. An OD 260/280 ratio over 1.8 is pre-ferred. Plasmids should be verified by agarose gel electropho-resis to confirm that the preparation is mostly in supercoiled form and free of RNA contamination. Use a supercoiled lad-der to verify plasmid sizes.14. Cell culture should be diluted in fresh medium a day or 2before transfection and allowed to grow to the desired density for optimal transfection. Avoid centrifuging cells the day of transfection as this can have a negative effect on transfection efficiency.15. The protocol describes production of lentiviral particles bytransient transfection of 20 mL suspension cultures. The pro-duction scale can be adjusted to fit other experimental require-ments as outlined in Note 4.16. The method has been successfully tested with different com-bination and number of plasmid constructs coding for lenti-virus vector. Therefore, the method can be easily adapted for lentivirus production with one preferred combination of plasmids after determination of optimal plasmid ratios.17. The final DNA concentration in culture medium is 1 m g/mLand the DNA to PEI mass ratio should be in the range of 1:3.These parameters should be kept constant for lentivirus vector production regardless of the production scale.18. The lentiviral transfer vector used in our studies carries afluorescent marker (GFP) that allows for assessment of the transfection efficiency using fluorescence microscopy or flow cytometry.19. It is recommended to perform an acetone test to verify thepacking efficiency and column integrity and a blank test prior to the run with running buffer.20. This method can be used to titer lentiviral vectors coding forGFP. It should be noted that the titration assay is basically the same as the traditional reporter gene expression assay with the exception that instead of using adherent target cells we have successfully employed suspension-growing target cells.No overnight cell attachment and trypsinization steps are required, thus, reducing the time and greatly simplifying the titration assay.21. Fixed samples can be stored in the dark at 4°C overnight.Formaldehyde has the double function of fixing the cells and inactivating the viral particles. Samples can be taken outside the contained laboratory for flow cytometry analysis after this step.。