全外显子组测序的具体方法及步骤
- 格式:docx
- 大小:55.10 KB
- 文档页数:1

全外显子捕获测序的杂交和封闭原理
做外显子捕获测序实验时,会用到Cot DNA,blocker(封闭接头序列),探针(DNA/RNA均可),链霉素磁珠等,它们具体起什么作用呢?下面,我们就来聊聊这个问题。
就实验原理来讲,外显子捕获测序的基本流程是:1.先对各个样品独立构建常规测序文库;2:等摩尔比例混合各个待杂交的文库;3:在杂交缓冲液体系内加入文库、杂交探针、Cot DNA、Blocker等;4.加入链霉素磁珠抓取杂交复合状态DNA;5:在洗脱缓冲液中洗脱目标文库,并扩增。
下图是正常建好的文库示意图。
大家都知道,DNA是双链互补配对的。
当加热打开DNA双链变性为单链DNA之后,慢慢降低温度,单链DNA会根据互补配对原则自然还原回双链状态(也就是退火过程)。
那么,可以想象一下,当很多不同的DNA文库混合在一起的时候,进行退火操作,可能会产生什么样子的DNA结合状态呢?下图说明了其中可能存在的3种情况。
由上图可知,不论哪种状态,都不可能捕获到目标DNA。
其实,情况2、3的杂交状中,P5或者P7端还可以杂交第三个、第四个等等分子。
所以,加入Blocker就是为了阻止P5、P7端的杂交,从而阻止非特异性杂交。
Cot DNA则是为了阻止原来互补配对的单链杂交回来。
带Biotin修饰的探针是为了杂交靶序列,链霉素磁珠是为了吸附Biotin修饰的杂交复合物。

全外显子测序报告解读原则与技巧全外显子测序是利用高通量测序技术对生物体全基因组外显子区域进行测序,从而揭示人类个体及群体基因组中与疾病相关的基因变异,是现代个性化医学的重要技术手段之一。
下面我们将介绍全外显子测序报告的解读原则和技巧。
解读原则:1.全面性:全外显子测序提供了全面、高通量的大量数据,必须对其进行全面、深入的解读。
同时需要结合临床资料,以全面、系统性的方式进行解读。
2.多参考性:全外显子测序可能会检测到一些变异,但并不一定与致病性相关。
因此,需要根据多个参考数据库、文献资料以及基于家系检测的疾病遗传性等多方面的数据进行判断和筛选。
3.个体化:全外显子测序报告需要与具体个体相关的临床资料、家族病史等进行结合,重点考虑与之相关的变异是否致病、临床意义何在等方面。
4.实用性:全外显子测序报告应当具有实用性,得出的结论应当能指导个体的诊断、治疗与遗传咨询。
解读技巧:1.对阳性结果进行验证:全外显子测序可能会检测到大量的单核苷酸多态性(SNP)、小的结构变异等,为了保证结果的精确性,最好对阳性结果进行验证,可以使用参考文献、数据库或其他现代检验技术进行检验。
2.避免过度解读:全外显子测序结果的解读需要考虑基因本身的复杂性,并非所有的变异都与疾病相关。
因此不应过度解读,需要根据科学方法进行分析和评价。
3.结合病史、家族史等临床资料:全外显子测序结果需要结合实际临床背景进行解读,包括基因检测的目的、临床表现、影响家庭、遗传风险等因素。
4.遵循实践指南:目前许多学会和机构都制定了全外显子测序报告解读的指南,如美国基因组医学协会(ACMG)和全球基因组联盟(GA4GH)等,解读应该遵循指南的原则和标准。
总之,全外显子测序是一项高复杂性的技术,其结果的解读需要谨慎,需要全面、深入地理解和分析,以确保结果的准确性和实用性。
同时,我们需要结合个体的临床信息和基因组数据来指导临床医生的决策和个体的诊断治疗方案,为个性化医学做出贡献。

全外显⼦组测序常见问题(上)1全外显⼦组测序必须要有参考基因组吗?必须有,如果没有参考因组,要提供近缘物种的序列,但不能保证捕获结果的可靠性。
因为捕获探针是根据提供的参考序列来设计的,如果已知⽬标区域与参考基因组⽐有较⼤出⼊,例如⼤⽚段的插⼊缺失,是不推荐的。
2为什么外显⼦测序在分析时需要跟全基因组⽐对,⽽不是直接与⽬标区域⽐对?第⼀,⽬标区域相对于全基因组⼀般较短,⽽且可能不连续,如果将⽬标区域单独提取出来,会影响区域边缘的序列⽐对效果;第⼆,⽆法评估捕获质量,例如脱靶率、on target⽐例等。
3全外显⼦组测序⼀般建议做多少倍的覆盖?⼀般做100×或150×。
较⾼的覆盖倍数,对于测异质性的遗传变质,可以发现⼩⽐例的突变。
另外,外显⼦测序的覆盖是随机的,这样较⾼的平均覆盖率有利于保证⼤部分的区域有⾜够的覆盖倍数。
4全外显⼦组测序深度的意义是什么?测序深度如何换算?测序深度代表了序列被探针组覆盖的次数,次数越⾼,测序结果的识别就越精确,后续的统计分析也就越准确。
如果做肿瘤、低频突变研究,建议测序深度⾄少应达到150×以上。
如果只看经典SNP、⾮低频突变,测序深度也⾄少应该在30×以上。
测序深度换算⽅法:⼀般⽬标区域的捕获效率在60-70%,安捷伦和罗⽒等外显⼦捕获试剂盒的⽬标区域⼤⼩在60Mb左右,即测序深度=10G*60%/60Mb=100×。
5全外显⼦组测序能够测出多⼤的⽚段缺失?⼤致能测出50bp的⽚段缺失。
由于外显⼦测序的覆盖很不平均,所以如果有⼤段的缺失,⽆法判断是因为杂交没有捕获到,还是因为缺失。
⽬前能够测到的,就是在⼀个read中发现的缺失。
⼀个read的长度也就是150bp,所以50bp以下的⽚段缺失可以从外显⼦测序中测出来。
6全外显⼦组测序可以做CNV分析吗?检测CNV的⽅法还有哪些?全外显⼦测序因为有⼀个杂交捕获的过程,这样就会有⼀个杂交捕获效率的问题。


针对外显子设计PCR测序引物教程在园子搜索后,没有看到长基因(大与1000base)最简洁方法,而我现在欧洲实验室里从事这方面工作,作了大量这方面的工作。
自乐不如同乐,愿将我们设计引物技巧与大家分享,敲字很辛苦,请斑竹给点分。
可能有战友说了,我们的长基因都是交给测序公司用鸟枪法来测全基因的。
当然,您有钱当然可以这样做。
我们的方法适用于基因测序筛查突变,步骤相对简便,比较经济。
另外,本实验室最近的一偏文章采用该法发在了NEMJ上,可见该法已经是经典成熟的。
(1)基础知识我们知道gDNA由非编码区,外显子,内含子构成。
我们关心的基因是否突变在非编码区,外显字以及临近外显子的一小段内含子上。
至于其他的内含子(gDNA中的大头),发生突变与否并不是我们关心的,其临床意义也相当小。
因此我们只要设计引物来PCR上面三个重点区域就可以了。
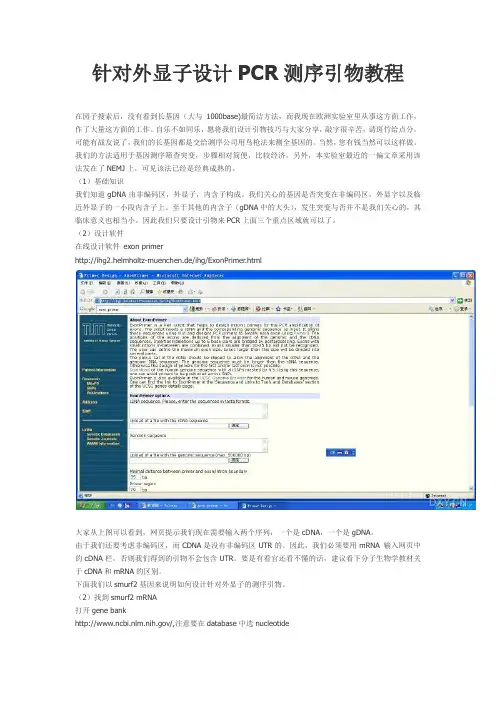
(2)设计软件在线设计软件exon primerhttp://ihg2.helmholtz-muenchen.de/ihg/ExonPrimer.html大家从上图可以看到,网页提示我们现在需要输入两个序列,一个是cDNA,一个是gDNA。
由于我们还要考虑非编码区,而CDNA是没有非编码区UTR的。
因此,我们必须要用mRNA 输入网页中的cDNA栏。
否则我们得到的引物不会包含UTR。
要是有看官还看不懂的话,建议看下分子生物学教材关于cDNA和mRNA的区别。
下面我们以smurf2基因来说明如何设计针对外显子的测序引物。
(2)找到smurf2 mRNA打开gene bank/,注意要在database中选nucleotide如下图蹦出一大串序列。
找到我们要的人类的smurf2Homo sapiens SMAD specific E3 ubiquitin protein ligase 2 (SMURF2), mRNA直接点我们要的序列名字,就得到了mRNA了,Format:GenBank FASTA Graphics More Formats选项中当然要求点选FASTA形式了把mRNA序列拖选,拷贝下来再拷贝入在线设计软件exon primer (见第一贴)http://ihg2.helmholtz-muenchen.de/ihg/ExonPrimer.html好了。


全外显子组测序的具体方法及步骤全外显子组测序(Whole Exome Sequencing,简称WES)是一种高通量测序技术,用于测定一个个体的所有外显子区域的DNA序列。
外显子是编码蛋白质的基因组区域,占据了人类基因组的约1-2%。
WES可以用于寻找致病基因突变,特别是在遗传性疾病的分子诊断中有广泛的应用。
下面将详细介绍WES的具体方法及步骤。
1.样品准备:-提取DNA:从待测个体的外周血或组织样品中提取总DNA。
-细胞裂解:使用特定组织裂解缓冲液将细胞或组织样品裂解,释放DNA。
-纯化DNA:通过离心等步骤,去除杂质,纯化DNA。
2.外显子库建立:- 靶向捕获:使用外显子组富集探针(baits)将DNA中的外显子区域进行加权,并去除非外显子区域的DNA片段。
-杂交反应:将靶向探针与DNA样品进行杂交反应,使探针与待测DNA的外显子区域发生特异性结合。
-洗涤:将未结合的探针洗掉,保留结合的外显子区域DNA片段。
-PCR扩增:对靶向捕获得到的DNA片段进行PCR扩增,以增加样品中外显子区域的DNA原料。
3.高通量测序:-数据库构建:将PCR扩增得到的外显子DNA片段建立一个DNA文库,用于测序。
- 测序反应:使用高通量测序平台(如Illumina HiSeq X)进行DNA文库的测序,得到大量的短序列片段(reads)。
- 数据处理:通过对这些reads进行去除低质量序列、比对到参考基因组等处理,获得高质量的测序数据。
4.数据分析:- 变异检测:使用专门的变异检测软件对样品中的变异进行分析,包括单核苷酸多态性(Single Nucleotide Polymorphisms,简称SNPs)和小片段插入缺失等。
-数据解读:将检测到的变异与公开的数据库进行对比,筛选出可能与疾病相关的变异。
-功能注释:对筛选出的变异进行功能注释,评估其潜在影响,进一步缩小候选基因的范围。
- 候选基因验证:对最终候选基因进行进一步的实验验证,如Sanger测序。

全外显子测序结题报告客户项目编号上海祥音生物科技有限公司目录1.项目概述 (3)2.建库测序流程 (3)2.1.DNA样本检测 (3)2.2.建库捕获 (3)2.3.库检及上机测序 (3)3.数据分析 (4)3.1.分析流程 (4)3.2.数据库信息 (4)3.3数据分析软件 (5)4.分析结果 (5)4.1.原始数据 (5)4.2.质量控制 (7)4.3.质量评估 (9)4.4.重要指标统计 (15)4.5变异检测结果 (21)5.参考文献 (28)1.项目概述2.建库测序流程2.1.DNA样本检测对DNA样品的检测主要包括3种方法:(1)琼脂糖凝胶电泳分析DNA降解程度以及是否有RNA及蛋白等污染。
(2)Nanodrop检测DNA的纯度(OD260/280比值)。
(3)Qubit对DNA浓度进行精确定量。
一般OD值在1.8~2.0之间,含量在1.5ug以上的DNA样品用来建库结果更好。
2.2.建库捕获Agilent SureSelect Human All Exon V6是 Agilent自主研发的全外显子捕获芯片,该产品汲取多个权威数据库(如RefSeq,OMIM_cds)的核心内容,具有更大的捕获区间,更高的捕获效率,保证外显子编码区的高覆盖率及SNP检出率。
将全外显子区域进行捕获并富集后,使用主流的 illumina、Life Technology 等测序平台进行高通量测序。
实验严格按照生产流程进行操作。
2.3.库检及上机测序1)取1μl文库使用Qubit dsDNA HS Assay Kit进行定量,记录文库浓度,文库浓度约在1-10ng/μl;2)取1μl样品使用Agilent2100Bioanalyzer system(Agilent DNA1000Kit)进行文库片段长度测定,文库长度约在220-320bp之间;3)使用高通量测序平台进行测序。
3.数据分析3.1.分析流程获得原始测序序列(Sequenced Reads)后,在有参考序列或参考基因组(GRCh37/hg19)的情况下,进行信息分析流程,大致包括以下两个部分:1)测序数据质量评估:主要对数据量、碱基质量、比对率、覆盖率、捕获率、均一性等指标进行统计,评估建库测序是否达到了标准,符合标准则进行后续分析。

ACMG全外显子测序指南摘要:美国医学遗传学与基因组学学会(ACMG)以前为序列突变的解释提供了指导.1在过去十年中,随着高通量测序的出现,测序技术迅速发展。
通过采用和利用下一代测序,临床实验室正在进行基因分型,单基因,基因组,外显子,基因组,转录组和遗传疾病表观遗传学检测的不断增加的遗传检测目录。
由于复杂性增加,基因检测的这种转变伴随着序列解释的新挑战。
在这方面,ACMG于2013年召集了一个由ACMG,分子病理学协会(AMP)和美国病理学家学会的代表组成的工作组,重新审视和修订了序列突变解释的标准和准则。
该组由临床实验室主任和临床医生组成。
本报告代表ACMG,AMP和美国病理学家利益相关者联盟组成的工作组的专家意见。
这些建议主要适用于临床实验室使用的遗传检测的范围,包括基因分型,单基因,panel,外显子和基因组。
本报告建议使用具体的标准术语- “致病性”,“可能致病性”,“不确定性意义”,“可能良性”和“良性”来描述在导致孟德尔病症的基因中鉴定的突变。
此外,该建议描述了基于使用典型类型的突变证据(例如,群体数据,计算数据,功能数据,分离数据)的标准将突变分类为这五个类别的过程。
由于本报告中描述的临床基因检测的分析和解释的复杂性增加,ACMG强烈建议临床分子遗传学检测应在经过临床实验室改进修订批准的实验室进行,结果由相关职业认证的临床分子遗传学家或分子遗传病理学家或同等学科专家进行解释。
关键词:ACMG实验室指导; 临床遗传检测; 解释;报告; 序列变异术语;突变报告前言临床分子实验室正在不断增加检测的新的序列突变,因为在检测患者标本时不断发现大量与基因疾病相关的基因。
虽然一些表型与单个基因相关,但许多与多个基因相关。
我们对任何给定序列突变的临床意义的理解是循序渐进的,其范围从那些几乎肯定是疾病致病性突变到几乎肯定是良性的突变。
虽然以前的美国医学遗传学和基因组学会(ACMG)的建议提供了序列突变的解释类别和解释算法,但是这些建议没有提供定义的术语或详细的突变分类指南.1。

外显子捕获测序原理外显子捕获测序是一种高效的基因组测序方法,它可以选择性地富集目标基因组的外显子区域,并对其进行测序。
这种方法的原理是利用特异性探针或引物,将外显子区域与其他非编码区域分离开来,从而提高测序效率和准确性。
外显子是基因组中编码蛋白质的区域,占据了整个基因组的很小一部分。
然而,它们却承载着大部分与遗传疾病相关的突变。
因此,通过对外显子进行测序,可以更好地理解基因变异与疾病之间的关系。
外显子捕获测序的步骤如下:1. 设计探针或引物:根据目标基因组的外显子序列,设计一组特异性的探针或引物。
这些探针或引物可以与外显子区域的序列互补配对,并将其与其他非编码区域分离开来。
2. 捕获外显子:将设计好的探针或引物与待测样本中的DNA或RNA杂交。
这样,只有与外显子序列互补的探针或引物才能与样本中的外显子结合,形成稳定的杂交复合物。
3. 分离非特异性序列:通过洗涤步骤,将与非编码区域结合的探针或引物去除,只保留与外显子区域结合的探针或引物。
这样,就实现了对外显子的选择性富集。
4. 扩增和测序:对富集后的外显子区域进行扩增和测序。
扩增可以使用PCR等方法,将外显子序列扩增为足够数量的DNA片段。
然后,通过高通量测序技术对这些DNA片段进行测序,得到外显子的序列信息。
外显子捕获测序的优势在于可以高效地测序目标基因组的外显子区域,避免了对整个基因组进行测序的复杂性和高成本。
此外,外显子捕获测序还可以提高测序的深度和准确性,使得检测到的突变更加可靠。
外显子捕获测序在医学研究和临床诊断中具有广泛的应用。
它可以用于寻找与遗传疾病相关的突变,帮助科学家和医生更好地理解疾病的发生机制。
此外,外显子捕获测序还可以用于个体化医学,根据个体的基因组信息,为患者提供更精准的治疗方案。
外显子捕获测序是一种高效、准确的基因组测序方法。
通过选择性富集外显子区域,可以更好地理解基因变异与疾病之间的关系,为医学研究和临床诊断提供有力支持。
全基因组重测序和全外显子重测序技术流程全基因组重测序和全外显子重测序技术介绍•全基因组重测序和全外显子重测序是现代基因组学研究中常用的技术。
•这两种技术可以提供大量关于个体基因组的信息,有助于研究遗传变异和疾病相关基因。
全基因组重测序•全基因组重测序是指对个体的全部DNA进行测序。
•流程包括:DNA提取、文库构建、测序、数据分析。
•DNA提取:从样本中提取高质量的基因组DNA。
•文库构建:将提取的DNA进行加工处理,生成可以进行测序的文库。
•测序:采用高通量测序技术,对文库进行测序,获取序列信息。
•数据分析:对获得的序列数据进行质量控制、比对和变异检测等分析。
全外显子重测序•全外显子重测序是指对个体的外显子区域进行测序。
•外显子是编码蛋白质的基因区域。
•流程包括:DNA提取、文库构建、测序、数据分析。
•DNA提取:与全基因组重测序相同,从样本中提取高质量的基因组DNA。
•文库构建:将提取的DNA进行加工处理,生成可以进行测序的文库。
•测序:采用高通量测序技术,对文库进行测序,获取外显子序列信息。
•数据分析:对获得的外显子序列数据进行质量控制、比对和变异检测等分析。
应用领域•全基因组重测序和全外显子重测序广泛应用于人类遗传研究、疾病基因研究、个体基因组学和进化生物学等领域。
•这些技术可以帮助揭示基因组的结构和功能,发现与疾病相关的遗传变异。
结论•全基因组重测序和全外显子重测序技术的发展,为基因组学研究提供了强大的工具。
•这些技术的应用不断拓展,为理解人类和其他生物的基因组差异以及与疾病相关基因的发现提供有力支持。
外显子测序原理
外显子测序是一种基因组测序技术,它的原理是通过对DNA中编码蛋白质的外显子区域进行测序,来揭示个体基因组的遗传信息。
外显子是基因组中编码蛋白质的部分,相对于整个基因组来说,外显子只占据了很小的比例,但却包含了大部分人类疾病相关的变异。
外显子测序的原理主要包括以下几个步骤,DNA提取、文库构建、高通量测序和数据分析。
首先,从样本中提取DNA,然后将DNA片段通过特定的方法构建成文库,接着进行高通量测序,获取大量的DNA序列信息。
最后,利用生物信息学分析软件对测序数据进行分析,鉴定外显子区域的变异信息。
在DNA提取过程中,需要保证提取的DNA质量和纯度,以确保后续测序的准确性和可靠性。
文库构建是将DNA片段连接到载体上,形成文库,这一步骤的关键是要避免DNA片段丢失或错位。
高通量测序是通过高通量测序技术对文库中的DNA片段进行大规模并行测序,产生海量的DNA序列数据。
数据分析是将测序得到的原始数据进行质控、比对、变异检测等一系列生物信息学分析过程,最终得到外显子区域的变异信息。
外显子测序技术的应用非常广泛,它可以用于研究人类疾病的发病机制、遗传变异的关联分析、药物靶点的筛选等领域。
通过对外显子的测序,可以发现与疾病相关的致病基因突变,为疾病的早期诊断和个体化治疗提供重要的依据。
同时,外显子测序也可以用于研究种群遗传结构、演化过程和基因型-表型关联等基因组学研究领域。
总的来说,外显子测序技术以其高通量、高效率、高准确性的特点,成为了当前研究基因组学和临床遗传学的重要工具。
随着测序技术的不断发展和成熟,外显子测序的应用前景将会更加广阔,为人类健康和疾病治疗带来更多的机遇和挑战。
精准医疗中的基因测序技术的操作指南精准医疗作为一种基于个体基因组信息的医疗模式,正在逐渐改变传统的诊断和治疗方式。
而基因测序作为精准医疗的重要工具之一,正发挥着日益重要的作用。
本文旨在提供一份详细的基因测序技术操作指南,为精准医疗中的基因测序工作提供参考。
一、准备工作1. 选择合适的基因测序方法:根据研究或临床需求,选择合适的基因测序方法,包括全基因组测序(WGS)、全外显子组测序(WES)、RNA测序等。
2. 样品准备:收集并储存DNA或RNA样品,确保样品质量良好。
3. 样品文库构建:根据选定的测序方法,进行文库构建工作,包括DNA或RNA的片段化、连接适配体、PCR扩增等步骤。
二、基因测序仪操作1. 仪器预热:按照仪器操作手册预热测序仪,并确保仪器温度稳定。
2. 质控操作:进行测序质控操作,包括使用合适的质控样品进行测序运行前检测,确保仪器及测序试剂的质量符合要求。
3. 数据输出设置:根据需求进行数据输出设置,包括测序深度、序列长度等参数的设定。
4. 测序运行:根据样品类型和选用的测序方法,运行合适的测序模式,如单端测序、双端测序等。
三、数据分析与解读1. 数据质控与预处理:对生成的测序数据进行质控和预处理,包括去除低质量序列、去除接头序列、去除PCR重复等。
2. 序列比对:将预处理后的测序数据与参考基因组进行比对,获得序列比对结果。
3. 变异检测:根据比对结果,采用合适的算法进行变异检测,包括单核苷酸多态性(SNP)、插入缺失(Indel)等变异类型的检测。
4. 结果解读:根据变异检测结果,结合相关数据库和文献,进行结果解读和注释,确定与研究或临床目的相关的变异。
5. 功能注释和通路分析:对于已鉴定的变异,进行其功能注释和通路分析,评估其可能的生物学功能和潜在影响。
6. 报告撰写:根据分析结果,编写测序报告,包括样品信息、测序方法、数据分析流程、主要变异结果等。
四、结果应用1. 研究应用:将分析结果应用于科学研究,探索基因与疾病之间的关联,研究相关的分子机制及生物学功能。
⼀⽂读懂全外显⼦测序家系突变筛选策略最近⽼师和同学经常问针对外显⼦测序的家系遗传病如何进⾏突变筛选,今天⼩編就撰稿⼀篇,希望对⽼师和同学有所帮助,话不多说,直接看下⾯的⼲货。
⼩编碎语:DNA测序中从测序区域的⼤⼩分为全基因组重测序,全外显⼦测序,靶向区域测序。
全外显⼦⼤概占DNA全部碱基对的1%,即⼤概30M的碱基,⽬前⼤部分测序的全外数据量为10G,测序深度⼤概为100X-150x左右,不同的试剂盒导致不同的捕获效率的不同,不同试剂盒的均⼀度的不同导致不同区域实际深度不同,由于全外显⼦测序检测数据量适中(约10G),与全基因组相⽐(约90G),因为⼈类疾病有90%在外显⼦区域,全外显⼦测序⼗分具有性价⽐,可以发现与⼈类疾病关系密切的外显⼦部分的相关基因突变。
全外显⼦的测序应⽤⼗分⼴泛,整体从技术上来说,1)可以检测 SNV 的 germ line 突变;2)也可以在⼀定程度上检测肿瘤的 somatic 突变(深度200X以上);3)可以检测外显⼦区域的CNV, 融合等突变;从外显⼦测序技术延伸出的临床应⽤来说,可以应⽤于以下的⽅⾯:1)确定孟德尔遗传疾病相关基因;2)风险易感基因的发现(与全基因组关联分析类似);3)癌症相关研究(⾼深度情况下);全外显⼦测序的⾼通量分析流程如下:不同试剂盒导致捕获效率的不同,不同试剂盒均⼀度的区别导致不同区域实际深度差异,下表为⽬前市场上主流试剂盒的⽐较。
孟德尔遗传疾病相关研究(家系筛选)通过全外显⼦⽣物信息分析,通过初步将得到⼀些可能的致病突变;如果知道样本家系属于何种致病模式,可以使⽤不同的筛选模式进⾏筛选。
筛选模式有:1)常染⾊体隐性遗传甲、⼄:隐性遗传表现为双亲都没病,孩⼦患病。
患病个体亲代是突变携带者但表型正常,⼦代患病,如果不存在近亲结婚或⽣殖隔离等因素,往往患者同⼀致病基因的不同位点存在致病突变,即患者带有复合杂合突变(compound heterozygous mutations)。
全外显子组测序的具体方法及步骤
全外显子组测序(Whole Exome Sequencing,WES)是指利用序列捕获或者靶向技术将全基因组外显子区域DNA 富集后再进行高通量测序的基因组分析方法。
与全基因组重测序相比,全外显子组测序只需针对外显子区域的基因序列测序,覆盖度更深、数据准确性更高,更加简便、经济、高效。
技术优势
高性价,强分析,快速交付
外显子组测序主要用于识别和研究与疾病相关的编码区的基因组变异。
结合大量的公共数据库提供的外显子数据和正常人群数据库, 有利于更好地排除无害突变及解释变异信息之间的关联和致病机理。
技术路线
技术参数
样本要求。