配位化学讲义第四章(1)价键理论、晶体场理论(完整)
- 格式:pdf
- 大小:3.55 MB
- 文档页数:20



配位化学Coordination Compounds内容提要1.基本概念①配合物的定义②配合物的组成③配合物的命名2.化学键理论①配合物的价键理论②晶体场理论3.配位平衡①配位平衡常数②配位平衡的移动4.鳌合物和生物配体①鳌合效应②影响鳌合物稳定性的因素第一节配位化合的基本概念一、配位化合物的定义•配合物是以具有接受电子对的离子或原子(统称中心原子)为中心,与一组可以给出电子对的离子或分子(统称配体),以一定的空间排列方式在中心原子周围所组成的质点(配离子或配分子)为特征的化合物。
CuSO 4Solution adding NaOH Cu(OH)2Precipitation [Cu(NH 3)4]SO 4Complex第一节基本概念二、配合物的组成•多数配合物由配离子与带相反电荷的离子组成,•带正电荷的配离子称为配阳离子,带负电荷的配离子称为配阴离子,配合物也可以是电中性的配位分子,•含配离子的化合物和配位分子统称为配合物,•习惯上把配离子也称为配合物。
[Ag(NH 3)2]+; [HgI 4]2-; [Fe(NCS)4]-; Pt(NH 3)2Cl 2]第一节基本概念1.配合物的内层(inner sphere)和外层(outer sphere)[Cu ( NH 3 )4 ]SO 4Central Ligands (‘赖跟的)atomInner sphere Outer sphereCoordination compound电中性的配位分子只有内层,没有外层。
第一节基本概念2.中心原子(central atom)•配合物中接受孤对电子的阳离子或原子统称为中心原子。
•中心原子一般是金属离子,大多为过渡元素,特别是第ⅧB族元素以及相邻近的一些副族元素。
•某些副族元素的原子和高氧化值非金属元素的原子也是较常见的中心原子,如[Ni(CO)4]中的Ni(0)、[SiF6]2-中的Si(Ⅳ)第一节基本概念3.配体(ligand)和配位原子(ligating atom)•与中心原子以配位键结合的阴离子或中性分子称为配体[Ag(NH3)2]+中NH3、[Ni(CO)4]中CO 、[SiF6]2-中F-•配体中直接向中心原子提供孤对电子形成配位键的原子称为配位原子NH3中的N、CO中的C、F-中的F •配位原子的最外电子层都有孤对电子,常见的是电负性较大的非金属的原子N、O、C、S、F、Cl、Br、I第一节基本概念3.配体(ligand)和配位原子(ligating atom)单齿配体(monodentate ligand)多齿配体(multidentate ligand)•单齿配体NH3、H2O、F-、Cl-少数配体虽有两个配位原子,由于两个配位原子靠得太近,只能选择其中一个与中心原子成键,故仍属单齿配体-、ONO-、SCN-、NCS-如CN-、NC-、NO2第一节基本概念3.配体(ligand)和配位原子(ligating atom)•多齿配体¾双齿配体:H2N-CH2-CH2-NH2(乙二胺,简写为en)¾三齿配体:H2NCH2CH2NHCH2CH2NH2(二亚乙基三胺,简写为DEN) ¾六齿配体:乙二胺四乙酸根Ethylenediaminetetraacetic ion , EDTA C H 2C H 2N N H 2C C H 2H 2C C H 2C O -O C O -OC C O -O -O O第一节基本概念第一节基本概念4.配位数(coordination number)•配合物中直接与中心原子键合的配位原子数目。

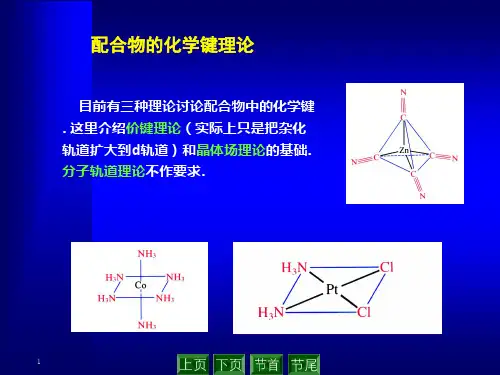
第三章配合物的化学键理论目标:解释性质,如配位数、几何结构、磁学性质、光谱、热力学稳定性、动力学反应性等。
三种理论:①价键理论、②晶体场理论、③分子轨道理论第一节价键理论(Valence bond theory)由L.Pauling提出要点:①配体的孤对电子可以进入中心原子的空轨道;②中心原子用于成键的轨道是杂化轨道(用于说明构型)。
一、轨道杂化及对配合物构型的解释能量相差不大的原子轨道可通过线性组合构成相同数目的杂化轨道。
对构型的解释(依据电子云最大重叠原理:杂化轨道极大值应指向配体)二、AB n型分子的杂化轨道1、原子轨道的变换性质考虑原子轨道波函数,在AB n分子所属点群的各种对称操作下的变换性质。
类型轨道多项式sp x xp p y yp z zd xy xyd xz xzd d yz yzd x2-y2x2-y2d z22z2-x2-y2(简记为z2)*s轨道总是按全对称表示变换的。
例:[HgI3]- (D3h群)平面三角形A1′:d z2、sE′:(p x、p y )、(d x2-y2、d xy)A2″:p zE″:(d xz、d yz)2、σ轨道杂化方案1)四面体分子AB4(Td)[CoCl4]2-以四个杂化轨道的集合作为分子点群(Td)表示的基,确定该表示的特征标:E 2 -1 2 0 0 (z2, x2-y2)T1 3 0 -1 1 -1T2 3 0 -1 -1 1 (xy,xz,yz) (x,y,z)a(A1)=1/24(1×4+8×1×1+3×1×0+6×1×0+6×1×2)=1a(A2)=1/24 [1×4+8×1×1+3×1×0+6×(-1)×0+ 6×(-1)×2]=0a(E)=1/24 [2×4+8×(-1)×1+3×2×0+6×0×0+ 6×0×2]=0a(T1)=1/24 [3×4+8×0×1+3×(-1)×0+6×1×0+6×(-1)×2]=0a(T2)=1/24 [3×4+8×0×1+3×(-1)×0+6×(-1)×0 +6×1×2]=1约化结果Γ=A1+T2由特征标表:A1T2s (p x、p y、p z)(d xy、d xz、d yz)可有两种组合:sp3(s、p x、p y、p z)、sd3(s、d xy、d xz、d yz)* 以一组杂化轨道为基的表示的特征标的简化计算规则:Γ 5 2 1 3 0 3约化结果:Γ= 2A1′+A2〞+E′A1′A2〞E′s p z (p x、p y)d z2(d xy、d x2-y2)两种可能的组合:(s、d z2、p z 、p x、p y)( s、d z2、p z、d xy、d x2-y2)约化得:Γ=A1g+B1g+E uA1g B1g E us d x2-y2(p x、p y)d z2两种类型:dsp2(d x2-y2、s、p x、p y)、d2p2(d z2、d x2-y2、p x、p y)5)八面体AB6(O h) 例:[Fe(H2O)6]3+(d z2、d x2-y2、s、p x、p y、p z) 3、π成键杂化方案在AB n分子中,原子A上要有2n个π型杂化轨道和在B原子上的2n个π原子轨道成键。



配位化学讲义第四章(1)价键理论、晶体场理论第三章配合物的化学键理论目标:解释性质,如配位数、几何结构、磁学性质、光谱、热力学稳定性、动力学反应性等。
三种理论:①价键理论、②晶体场理论、③分子轨道理论第一节价键理论(Valencebond theory)由L.Pauling提出要点:①配体的孤对电子可以进入中心原子的空轨道;②中心原子用于成键的轨道是杂化轨道(用于说明构型)。
一、轨道杂化及对配合物构型的解释能量相差不大的原子轨道可通过线性组合构成相同数目的杂化轨道。
对构型的解释(依据电子云最大重叠原理:杂化轨道极大值应指向配体)指向实例sp3、sd3杂化四面体顶点Ni(CO)4sp2、sd2、dp2、d3杂化三角形顶点[AgCl3]2-dsp2、d2p2 杂化正方形顶点[PtCl4]2-d2sp3杂化八面体顶点[Fe(CN)6]4-sp杂化直线型[AgCl2]-二、AB n型分子的杂化轨道1、原子轨道的变换性质考虑原子轨道波函数,在AB n分子所属点群的各种对称操作下的变换性质。
类型轨道多项式sp x xp p y yp z zd xy xyd xz xzd d yz yzd x2-y2x2-y2d z22z2-x2-y2(简记为z2)*s轨道总是按全对称表示变换的。
例:[HgI3]- (D3h群)平面三角形A1′:d z2、sE′:(p x、p y )、(d x2-y2、d xy)A 2″:p zE″:(d xz、d yz)2、σ轨道杂化方案1)四面体分子AB4(Td)[CoCl4]2-以四个杂化轨道的集合作为分子点群(Td)表示的基,确定该表示的特征标:r1r4r2r3恒等操作,χ(E)=4 C3操作,χ(C3)=1对C2、S4和σd用同样方法处理,得T d E 8C3 3C2 6S46σdΓ 4 1 00 2约化:T d E 8C3 3C2 6S4 6σdA1 1 1 1 11A2 1 1 1 -1 - 1E 2 -1 2 00 (z2, x2-y2)T1 3 0 -1 1 -1T2 3 0 -1 -11 (xy,xz,yz) (x,y,z)a(A1)=1/24(1×4+8×1×1+3×1×0+6×1×0+6×1×2)=1a(A2)=1/24 [1×4+8×1×1+3×1×0+6×(-1)×0+6×(-1)×2]=0a(E)=1/24 [2×4+8×(-1)×1+3×2×0+6×0×0+6×0×2]=0a(T1)=1/24 [3×4+8×0×1+3×(-1)×0+6×1×0+6×(-1)×2]=0a(T2)=1/24 [3×4+8×0×1+3×(-1)×0+6×(-1)×0+6×1×2]=1约化结果Γ=A1+T2由特征标表:A1T2s(p x、p y、p z)(d xy、d xz、d yz)可有两种组合:sp3(s、p x、p y、p z)、sd3(s、d xy、d xz、d yz)* 以一组杂化轨道为基的表示的特征标的简化计算规则:①不变(1)②改变符号(-1)③与其他函数变换(0)2)再以[CdCI5]3-三角双锥(D3h)为例:41325D3h E 2C33C2σh2S3 3σvΓ 5 2 13 0 3约化结果:Γ= 2A1′+A2〞+E′A1′A2〞E′s p z (p x、p y)d z2(d xy、d x2-y2)两种可能的组合:(s、d z2、p z 、p x、p y)( s、d z2、p z、d xy、d x2-y2)3)[HgI3]- ( D3h)123D3h E 2C3 3C2σh2S33σvΓ 3 0 13 0 1约化得:Γ=A1′+E′A1′E′s (p x、p y)d z2(d xy、d x2-y2)可能的组合有:(s、p x、p y)、(s、d xy、d x2-y2)、(d z2、p x、p y)、(d z2、d xy、d x2-y2)4)平面AB4型分子(D4h)例:[PtCl4]2-C2′C2″D4h E 2C4(C41,C43) C2(C42) 2C2′2C2″i 2S4σh 2σv2σdΓ 4 0 0 20 0 0 4 2 0约化得:Γ=A1g+B1g+E uA1g B1g E us d x2-y2(p x、p y)d z2两种类型:dsp2(d x2-y2、s、p x、p y)、d2p2(d z2、d x2-y2、p x、p y)5)八面体AB6(O h) 例:[Fe(H2O)6]3+O h E 8C3 6C26C4 3C2i 6S4′8S6 3σh 6σdΓ 6 0 0 2 2 0 0 0 4 2约化得:Γ=A1g+E g+T1u A1g E gT1us (d z2、d x2-y2) (p x、p y、p z)只有唯一的d2sp3杂化(d z2、d x2-y2、s、p x、p y、p z)3、π成键杂化方案在AB n分子中,原子A上要有2n个π型杂化轨道和在B原子上的2n个π原子轨道成键。


.配位化合物的价键理论配合物的晶体场理论一.配合物的构型与中心的杂化方式二中心杂化轨道的形成1. ns np nd 杂化1 个 4s 空轨道,3 个 4p 空轨道和2 个 4d 空轨道形成 sp3d2杂化轨道,正八面体分布。
6 个F-的 6 对孤对电子配入sp3d2空轨道中,形成正八面体构型的配合单元。
例 2 Ni(CO)4的成键情况在配体 CO 的作用下,Ni 的价层电子重排成 3d104s0形成 sp3杂化轨道,正四面体分布,4 个CO 配体与 sp3杂化轨道成配键,形成的 Ni(CO)4构型为正四面体。
例 1 和例 2 的相同点是,配体的孤对电子配入中心的外层空轨道, 即 ns np nd 杂化轨道, 形成的配合物称外轨型配合物. 所成的键称为电价配键. 电价配键不是很强.例 1 和例 2 的不同点是,CO 配体使中心的价电子发生重排,这样的配体称为强配体。
常见的强配体有 CO、 CN-、NO2-等;例1 中 F-不能使中心的价电子重排,称为弱配体。
常见的弱配体有 F-、Cl-、H2O 等。
而 NH3等则为中等强度配体。
对于不同的中心,相同的配体其强度也是不同的。
2. (n-1) d ns np 杂化例 3 讨论的成键情况形成 d2sp3杂化,使用 2 个 3d 轨道, 1 个 4s 轨道,3个4p 轨道。
用的内层 d 轨道。
形成的配离子为正八面体构型。
空出 1 个内层 d 轨道,形成 dsp2杂化轨道,呈正方形分布。
故构型为正方形。
例 3 和例 4 中,杂化轨道均用到了 ( n - 1 ) d 内层轨道,配体的孤对电子进入内层,能量低,称为内轨配合物,较外轨配合物稳定。
所成的配位键称为共价配键。
三价键理论中的能量问题内轨配合物稳定,说明其键能 E内大,大于外轨的 E外,那么怎样解释有时要形成外轨配合物呢?其能量因素如何?上面的例题中我们看到,形成内轨配合物时发生电子重排,使原来平行自旋的 d 电子进入成对状态,违反洪特规则,能量升高。

第三章配合物的化学键理论目标:解释性质,如磁学性质、光谱等。
三种理论:①价键理论、②晶体场理论、③分子轨道理论第一节价键理论(Valence bond theory)由L.Pauling提出要点:①配体的孤对电子可以进入中心原子的空轨道;②中心原子用于成键的轨道是杂化轨道(用于说明构型)。
一、轨道杂化及对配合物构型的解释对构型的解释(依据电子云最大重叠原理:杂化轨道极大值应指向配体)指向实例sp3、sd3杂化四面体顶点Ni(CO)4sp2、sd2、dp2、d3杂化三角形顶点[AgCl3]2-dsp2、d2p2 杂化正方形顶点[PtCl4]2-d2sp3杂化八面体顶点[ Fe(CN)6]4-二、对配合物磁性的解释1)配合物磁性与配合物中成单电子数的关系配合物的分子磁矩μ与配合物中未成对电子数n 有关。
如:对某些配合物:µ=[n(n+2)]1/2 B.M.1B.M. = 9.27×10-21erg·G-12)实验发现:如:K4[Fe(CN)6] µ=0.00 B.M.FeSO4.7H2Oµ=5.10 B.M.②内、外轨型配合物磁性③继承了传统的价键概念(配位共价键),简明易于理解。
2)不足A. 定量程度差。
B. 无法说明Cu2+平面正方形内轨型配合物的稳定性如[Cu(NH3)4]2+:要点:①把配体视为点电荷或偶极子(不考虑其结构);②配体与中心离子间的作用是纯静电相互作用,不存在电子交换,即不形成任何共价键。
二、d轨道能级分裂(单电子能级的分裂)1、定义:由于d轨道空间取向不同,与非球形对称静电场的作用则不相同,引起d轨道能级发生分裂。
Δt = E t2 - E e = =4/9Δo 40/9Dq---------(1)同理,若选Es为能量零点,则3E t2+2E e=5E s=0---------(2)联立(1)和(2),解出:E t2=1.78Dq,E e=-2.67D q三、d电子排布及配合物磁性4、四面体配合物由于Δt=4/9Δo, 大多为弱场高自旋排布。